What Are Histone Modifications
Histones are basic proteins found in the chromatin of eukaryotic cells. Based on their amino acid composition, histones are categorized into two classes: lysine-rich (H1, H2A, and H2B) and arginine-rich (H3 and H4) (Figure 1). Their abundance of basic amino acids enables histones to interact with DNA, forming the nucleosome structure.
The nucleosome is the fundamental structural unit of the chromosome. Each nucleosome consists of a core octameric complex of histones H2A, H2B, H3, and H4, around which 147 base pairs of DNA are wrapped. Additional histones, H1 and H5, serve as linkers between adjacent nucleosomes, stabilizing the DNA between them.
Histone modification refers to the process by which histones undergo various chemical modifications such as methylation, acetylation, and phosphorylation under the action of specific enzymes. Each histone structure extends a small segment, known as the N-terminal tail, which protrudes from the core structure and is the primary site for these post-translational modifications (PTMs). Among the histones, H3 is the most frequently modified.
Numerous types of histone modifications have been identified. Well-studied modifications include acetylation, methylation, phosphorylation, and ubiquitination. Other modifications such as glycosylation, carbonylation, ADP-ribosylation, adenylylation, succinylation, 2-hydroxyisobutyrylation, biotinylation, formylation, malonylation, glutarylation, β-hydroxybutyrylation, N-acetylglucosaminylation, citrullination, crotonylation, and isomerization require further investigation.
Each modification is mediated by a specific set of enzymes that add or remove chemical groups to or from the amino acid residues of histones. Methylation is considered the most stable form of histone modification, making it suitable for conveying stable epigenetic information. In contrast, acetylation is highly dynamic, and other modifications such as phosphorylation, adenylylation, ubiquitination, and ADP-ribosylation are also less stable (Figure 2).
These modifications can occur in various combinations, each exerting distinct biological functions. Modifications of histones lead to alterations in chromatin conformation, thereby influencing the interactions between DNA and histones, ultimately regulating gene transcription. Consequently, histone PTMs, along with DNA methylation, constitute fundamental mechanisms for gene and epigenetic regulation, playing vital roles in cellular and developmental processes.
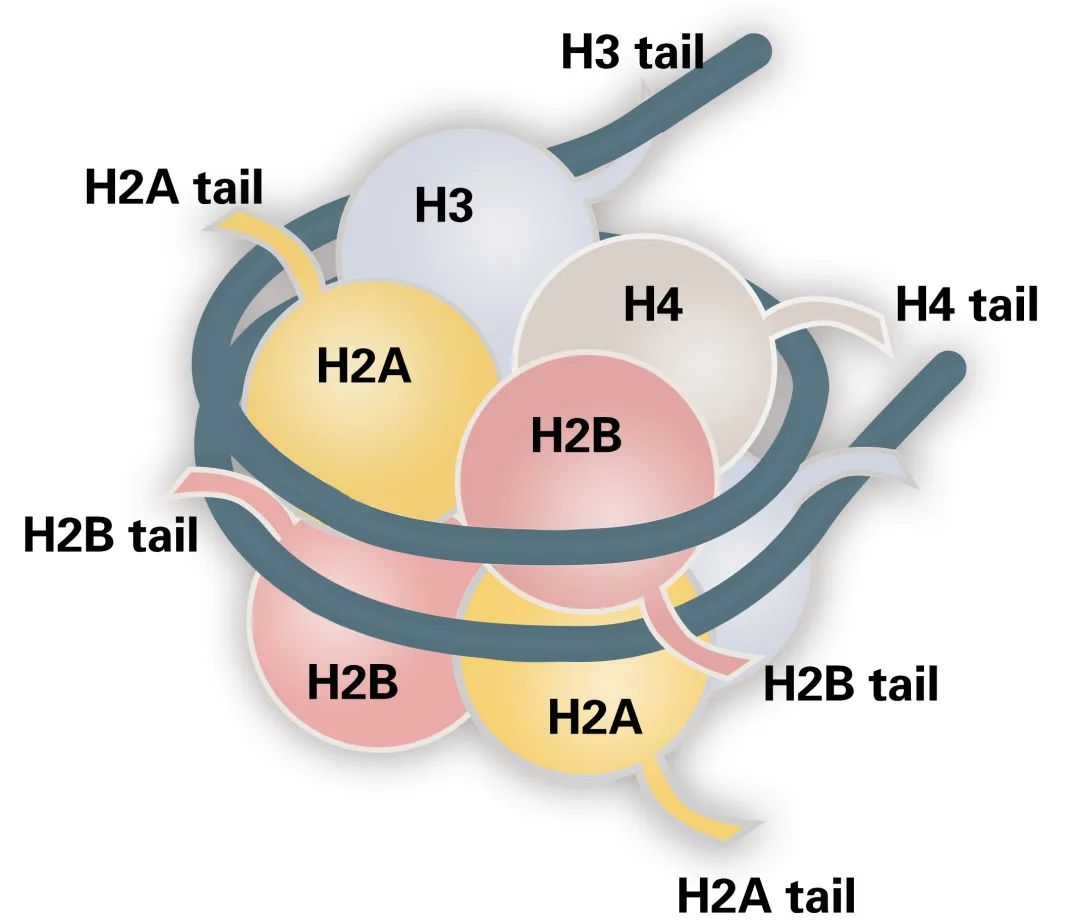 Figure 1 The structure of the nucleosome
Figure 1 The structure of the nucleosome
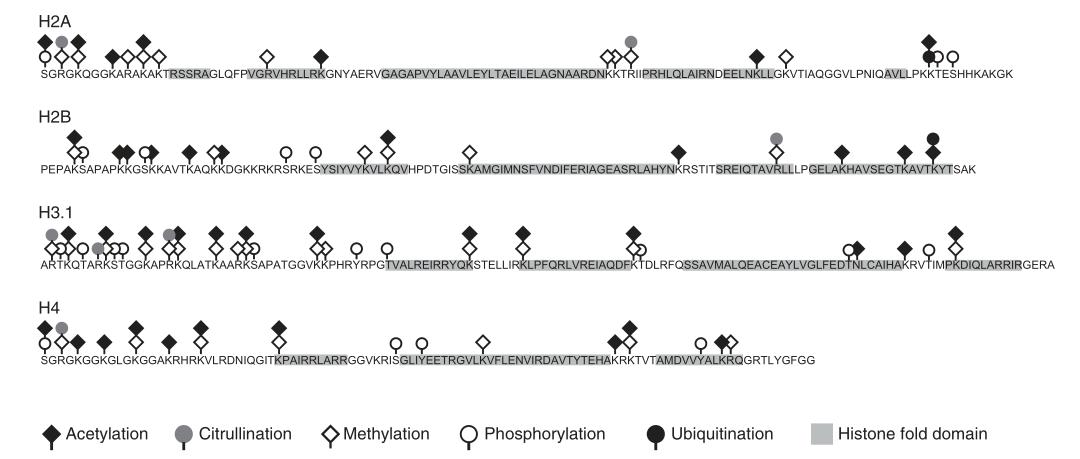 Figure 2 Common post-translational modifications of core histones
Figure 2 Common post-translational modifications of core histones
Histone Modifications and Modifying Enzymes
During the mid-20th century, significant advancements were made in the study of histone modifications. Allfrey et al. (1964) postulated a hypothesis indicating a positive correlation between the levels of histone acetylation and methylation and the activation of gene transcription. This hypothesis has been extensively substantiated by subsequent research, which also identified other types of histone modifications, such as phosphorylation and ubiquitination, involved in transcriptional regulation in Arabidopsis (Ueda and Seki, 2020). Among these modifications, histone acetylation and methylation have been particularly well-studied and are recognized as critical epigenetic regulatory mechanisms in gene expression.
Histone modifications are reversible covalent modifications whose occurrence, removal, and functional implications are meticulously regulated by specific histone-modifying enzymes and their associated cofactors (Figure 3). These enzymes are categorized into three primary classes: Writers, Erasers, and Readers/Effector proteins (Liu et al., 2010). Writer enzymes facilitate the addition of chemical groups to histones, effectuating the modification. Representative Writer enzymes include histone acetyltransferases (HATs), histone methyltransferases (HMTs), kinases, and ubiquitin ligases.
In contrast, Eraser enzymes are responsible for removing these modifications from histones. Examples of Eraser enzymes encompass histone deacetylases (HDACs), histone demethylases (HDMs), phosphatases, and deubiquitinases. Reader proteins recognize and specifically bind to substrates with particular post-translational modifications, thereby mediating downstream biological effects.
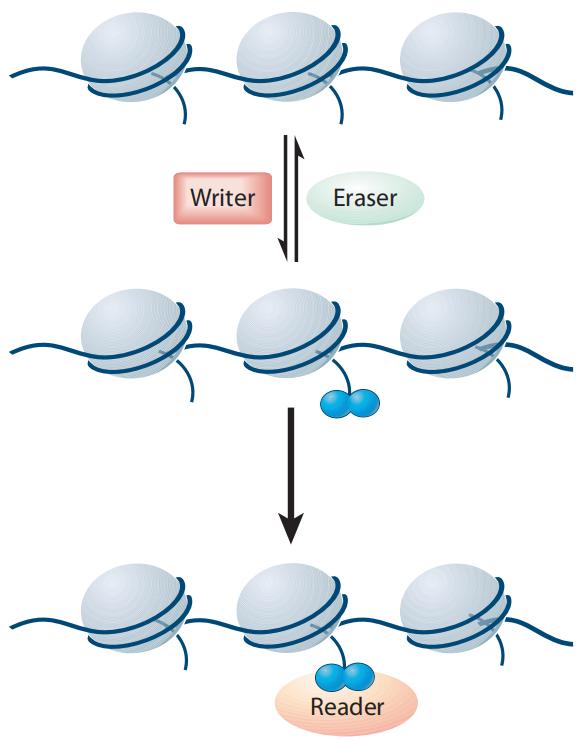 Figure 3 Writers, Erasers, and Readers/Effectors jointly regulate the levels and types of histone modifications (Liu et al., 2010).
Figure 3 Writers, Erasers, and Readers/Effectors jointly regulate the levels and types of histone modifications (Liu et al., 2010).
Histone Acetylation Modifications
Sites and Functions of Histone Acetylation
Histone acetylation, a prevalent post-translational modification (PTM) of histones, plays a critical role in the epigenetic regulation of gene expression in eukaryotic cells. Similar to other PTMs, the acetylation process is reversible, with its levels dynamically regulated by the coordinated action of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Figure 4). These processes maintain a dynamic equilibrium in vivo, collectively modulating gene transcription. Typically, histone acetylation facilitates gene activation, whereas deacetylation exerts a repressive effect on gene expression.
Histone acetylation predominantly occurs on specific lysine residues at the N-terminal tails of histones, including H3K9 (lysine 9 on histone H3), H3K14, H3K27, H3K36, H4K5, H4K8, H4K12, and H4K16, with H3K27 being particularly well-studied. Research indicates that H3K27ac is primarily localized at promoters and enhancers of actively transcribed genes, often co-occurring with H3K4me3 to synergistically promote gene activation. Additionally, H3K27ac can form super-enhancers in intergenic regions, further enhancing gene expression.
Recent studies have demonstrated the critical involvement of histone acetylation in both plant physiology and human pathologies. For instance, H3K9ac plays a pivotal role in poplar under drought stress, while H3K27ac is implicated in the pathogenesis of leukemia. Moreover, several activators and inhibitors of HATs and HDACs have been approved for the treatment of certain cancers, underscoring their potential therapeutic value in medical applications.
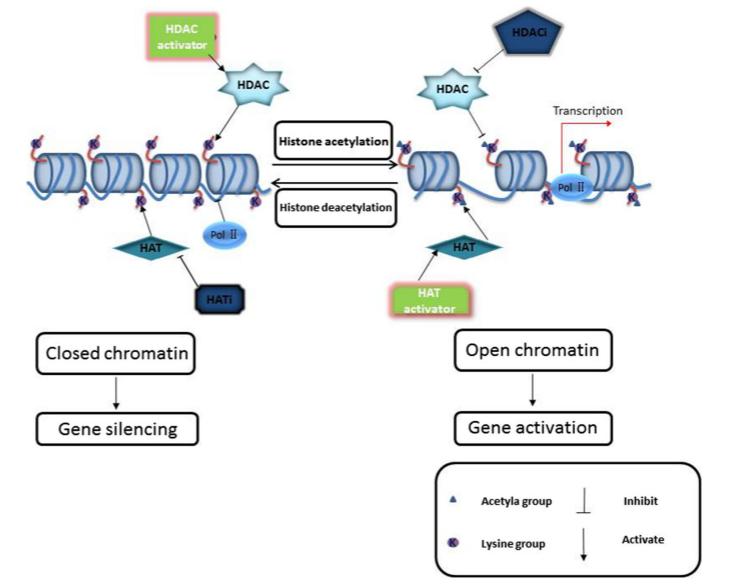 Figure 4 Histone acetylation and deacetylation
Figure 4 Histone acetylation and deacetylation
Table1 : Histone Acetylation Modification Types and Functions
| Histone Acetylation Modification |
Function |
| H3K9ac |
Transcription activation |
| H3K14ac |
Upregulation of gene expression such as TNFa, IL-6 |
| H3K18ac |
Transcription activation |
| H3K27ac |
Transcription activation |
| H3K56ac |
Activation |
| H4K5ace |
Approximately one-third of genes exhibit high levels of H4K5ac; about 65% coexist with H4K12ac |
| H4K8ac |
Controls transcription of ncRNA via the KAT2B-H4K8ac-BRD4 axis |
| H4K12ac |
Plays a crucial role in regulating gene expression during early embryonic development |
| H4K16ac |
Regulates the transition from heterochromatin to euchromatin; prepares genes for activation by modulating nucleosome accessibility prior to transcription activation |
Histone Acetyltransferases
Histone acetyltransferases (HATs) represent a supergene family, with over 20 distinct types identified to date. Based on structural characteristics, plant HATs can be categorized into four principal families: GNAT (Gcn5-related N-acetyltransferase), MYST (MOZ-YBF2/SAS3-SAS2-TIP60), CBP/p300 (CREB-binding protein), and TAFII-250 (TATA-binding protein-associated factor) (Lee and Workman, 2007). Each HAT family exhibits specificity for acetylating different lysine residues on histones: GNAT primarily targets H3K14 or H3K12, MYST focuses on H4K5, and CBP/p300 demonstrates broader activity, capable of acetylating all lysine residues subject to acetylation.
The GNAT family is the largest among HATs, encompassing members such as GCN5, PCAF, Elp3, Hat1, and Hpa2. In plant GNAT family proteins, the N-terminal region contains a conserved sequence exceeding 100 amino acids, known as the HAT domain, while the C-terminal region houses a bromodomain. The HAT domain comprises four motifs (C, D, A, and B), with the A motif being highly conserved and responsible for binding acetyl-CoA. The bromodomain interacts with acetylated lysine residues on histone tails, a mechanism critical for chromatin structure modulation and gene expression regulation (Lee and Workman, 2007).
Histone Deacetylases
Histone deacetylases (HDACs) exhibit greater diversity and number compared to histone acetyltransferases (HATs). Currently identified HDACs can be categorized into three major subfamilies: RPD3/HDA1, HD2, and SIR2. The RPD3/HDA1 subfamily is widely distributed across various eukaryotic organisms. In contrast, the SIR2 subfamily, distinguished by its unique structure, shows no homology with other HDAC types. Notably, the HD2 subfamily is specific to plants.
Histone Methylation
Methylation Sites and Functions
Histone methylation, in contrast to histone acetylation, does not alter the charge of histones and does not directly affect histone-DNA interactions. Instead, the presence of methylation primarily influences the binding affinity of reader proteins, which subsequently modulates chromatin structure and leads to either transcriptional repression or activation (Liu et al., 2010). The regulatory outcome is contingent upon the specific amino acid residue undergoing methylation and the degree of methylation.
Methylation predominantly occurs on lysine (K) and arginine (R) residues within histones. Lysine residues can undergo mono-, di-, or trimethylation, denoted as me1, me2, and me3, respectively. Histone methylation exhibits dual regulatory functions in transcription, either activating or repressing gene expression. Generally, methylation of H3K4, H3K36, and H3K79 is associated with transcriptional activation, whereas methylation of H3K9, H3K27, and H4K20 is linked to transcriptional repression. However, the functional impact also depends on the number of methyl groups at the same site. For instance, H3K27me1 is associated with transcriptional activation, while H3K27me3 is linked to transcriptional repression.
Extensive research has demonstrated that histone methylation plays a critical role in the pathogenesis of various diseases. Consequently, it serves as a valuable marker for disease diagnosis, prognosis, and therapeutic development.
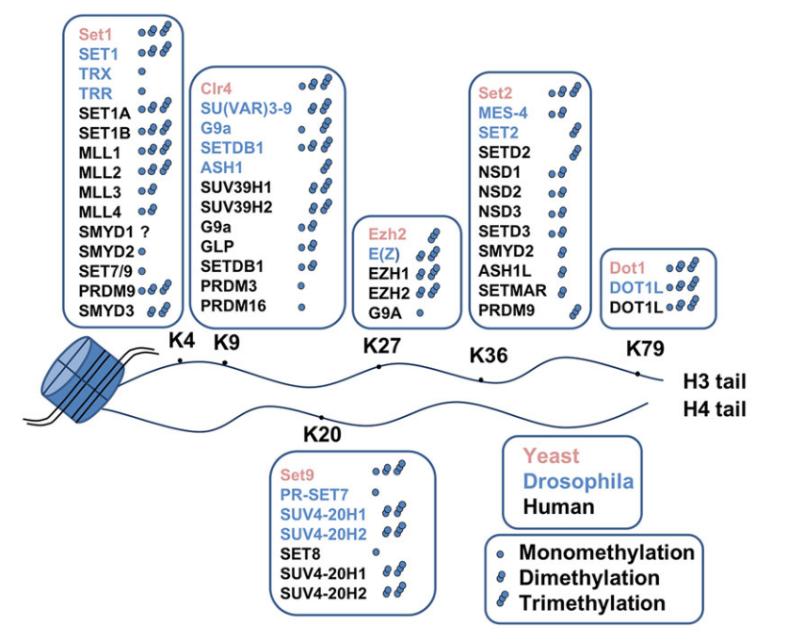 Figure 5 Histone methyltransferases and their lysine target sites on histones
Figure 5 Histone methyltransferases and their lysine target sites on histones
Table2 : Histone Methylation Modification Types and Functions
| Histone Methylation Modification |
Function |
| H3K4 |
Activation |
| H3K36 |
Activation |
| H3K79 |
Activation |
| H3K9 |
Inhibition |
| H3K27 |
Inhibition |
| H4K20 |
Inhibition |
| H3K27me1 |
Activation |
| H3K27me3 |
Inhibition |
| H3K4me1 |
Enrichment in enhancer regions |
| H3K4me2 |
Transcription of genes related to protein synthesis and protein homeostasis in DNA damage repair processes |
| H3K9me2 |
Associated with gene repression and heterochromatin formation |
| H3K9me3 |
Associated with gene repression and heterochromatin formation |
Histone Methyltransferases
Histone methyltransferases (HMTs) catalyze the transfer of a methyl group from S-adenosylmethionine (SAM or AdoMet) to specific lysine or arginine residues on histones, thereby mediating histone methylation (Figure 5). HMTs are categorized into two classes: histone lysine methyltransferases (HKMTs) and protein arginine methyltransferases (PRMTs) (Figure 6) (Liu et al., 2010).
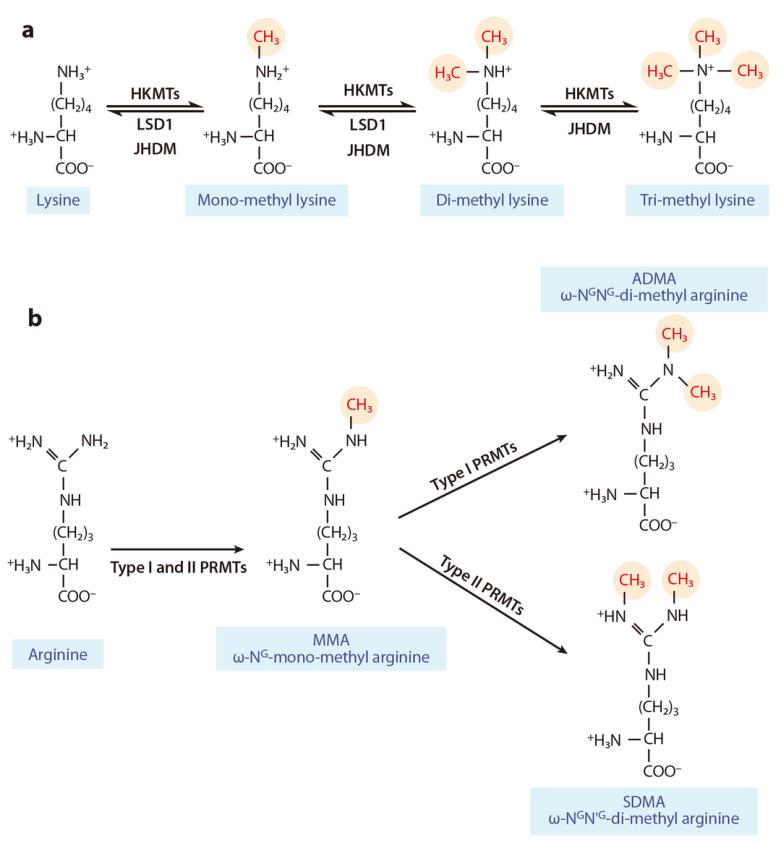 Figure 6 illustrates the complex and dynamic epigenetic regulation system of histone methylation and demethylation on lysine or arginine residues (Liu et al., 2010). (a) Mono-, di-, and trimethylation are catalyzed by histone lysine methyltransferases (HKMTs) and histone demethylases (LSD1, JHDM). (b) Type I and type II protein arginine methyltransferases (PRMTs) catalyze asymmetric and symmetric dimethylation, respectively. Symbols: ω-NG-mono-methyl arginine (MMA): Monomethylation of arginine; ω-NG,NG-di-methylarginine (ADMA): Asymmetric dimethylation of arginine; ω-NG,N'G-di-methylarginine (SDMA): Symmetric dimethylation of arginine.
Figure 6 illustrates the complex and dynamic epigenetic regulation system of histone methylation and demethylation on lysine or arginine residues (Liu et al., 2010). (a) Mono-, di-, and trimethylation are catalyzed by histone lysine methyltransferases (HKMTs) and histone demethylases (LSD1, JHDM). (b) Type I and type II protein arginine methyltransferases (PRMTs) catalyze asymmetric and symmetric dimethylation, respectively. Symbols: ω-NG-mono-methyl arginine (MMA): Monomethylation of arginine; ω-NG,NG-di-methylarginine (ADMA): Asymmetric dimethylation of arginine; ω-NG,N'G-di-methylarginine (SDMA): Symmetric dimethylation of arginine.
Histone Lysine Methyltransferases
Current knowledge identifies histone lysine methyltransferases (HKMTs) that contain a conserved SET domain (Su(var)3-9, Enhancer of zeste, and Trithorax) within the SET domain group (SDG) proteins, and those that lack the SET domain, such as Disruptor of Telomeric Silencing-1p (DOT1p) and Disruptor of Telomeric Silencing 1-like (DOT1L) proteins. Despite differences in their methyltransferase domains, both types utilize S-adenosylmethionine (SAM) as a methyl donor. To date, only SDG proteins have been recognized as HKMTs in plants. The plant SDG proteins constitute a large family. Based on previous studies, Li Huang et al. classified the SDG proteins in Arabidopsis thaliana and Oryza sativa into seven subfamilies (I-VII): E(z), Ash, Trx, ATXR5/6, Suv, SMYD, and SETD (Zhou et al., 2020) (Figure 7).
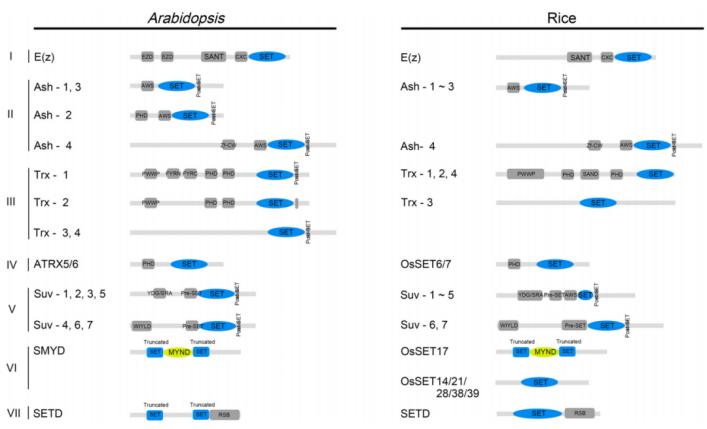 Figure 7 depicts the conserved domains of Arabidopsis thaliana and rice SET DOMAIN GROUP (SDG) proteins (Zhou et al., 2020). These domains include EZD (E(z) domain), SANT (SWI3, ADA2, N-CoR, and TFIIIB DNA-binding domain), CXC domain (rich in cysteine), AWS domain (associated with SET region), PHD (plant homeodomain), Zf-CW (zinc finger protein with conserved cysteine and tryptophan residues), PWWP (conserved Pro-Trp-Trp-Pro motif), FYRN (rich in F/Y residues at the N-terminus), FYRC (rich in F/Y residues at the C-terminus), YDG (conserved Tyr-Asp-Gly motif), SRA (a conserved SET and a RING zinc finger-associated domain), and RSB (ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) LSMT substrate binding domain).
Figure 7 depicts the conserved domains of Arabidopsis thaliana and rice SET DOMAIN GROUP (SDG) proteins (Zhou et al., 2020). These domains include EZD (E(z) domain), SANT (SWI3, ADA2, N-CoR, and TFIIIB DNA-binding domain), CXC domain (rich in cysteine), AWS domain (associated with SET region), PHD (plant homeodomain), Zf-CW (zinc finger protein with conserved cysteine and tryptophan residues), PWWP (conserved Pro-Trp-Trp-Pro motif), FYRN (rich in F/Y residues at the N-terminus), FYRC (rich in F/Y residues at the C-terminus), YDG (conserved Tyr-Asp-Gly motif), SRA (a conserved SET and a RING zinc finger-associated domain), and RSB (ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) LSMT substrate binding domain).
PRMTs can be categorized into two classes based on their catalytic activity. The first class, comprising PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8, facilitates the formation of monomethylated and asymmetrically dimethylated arginine residues (Figure 6). The second class, which includes PRMT5, PRMT7, and PRMT9, catalyzes the formation of monomethylated and symmetrically dimethylated arginine residues. Among the identified enzymes, PRMT1, PRMT2, PRMT4, PRMT5, PRMT6, and PRMT7 can methylate histones, whereas PRMT3, PRMT8, and PRMT9 primarily methylate non-histone substrates.
Histone Demethylases
Histone demethylases (HDMs) play pivotal roles in regulating the homeostasis of histone methylation. HDMs can be classified into two categories based on their mechanisms: lysine-specific demethylase 1 (KDM1/LSD1) and Jumonji C domain-containing demethylases (JMJ). KDM1/LSD1 catalyzes demethylation of mono- and dimethylated lysine residues through a flavin adenine dinucleotide (FAD)-dependent amine oxidation reaction. Conversely, the demethylation reaction catalyzed by JMJ proteins is a dioxygenase reaction dependent on iron (II) and α-ketoglutarate. JMJ proteins play crucial roles in histone demethylation, removing methyl modifications on H3K4, H3K9, H3K27, and H3K36.
Arabidopsis thaliana and Oryza sativa possess 21 and 20 Jumonji C domain-containing proteins (JMJs), respectively. Luo et al. (2013) classified these JMJs into five subfamilies based on protein sequence similarity (Figure 8B): KDM5/JARID1 group, KDM4/JHDM3 group, KDM3/JHDM2 group, JMJD6 group, and JmjC domain-only group.
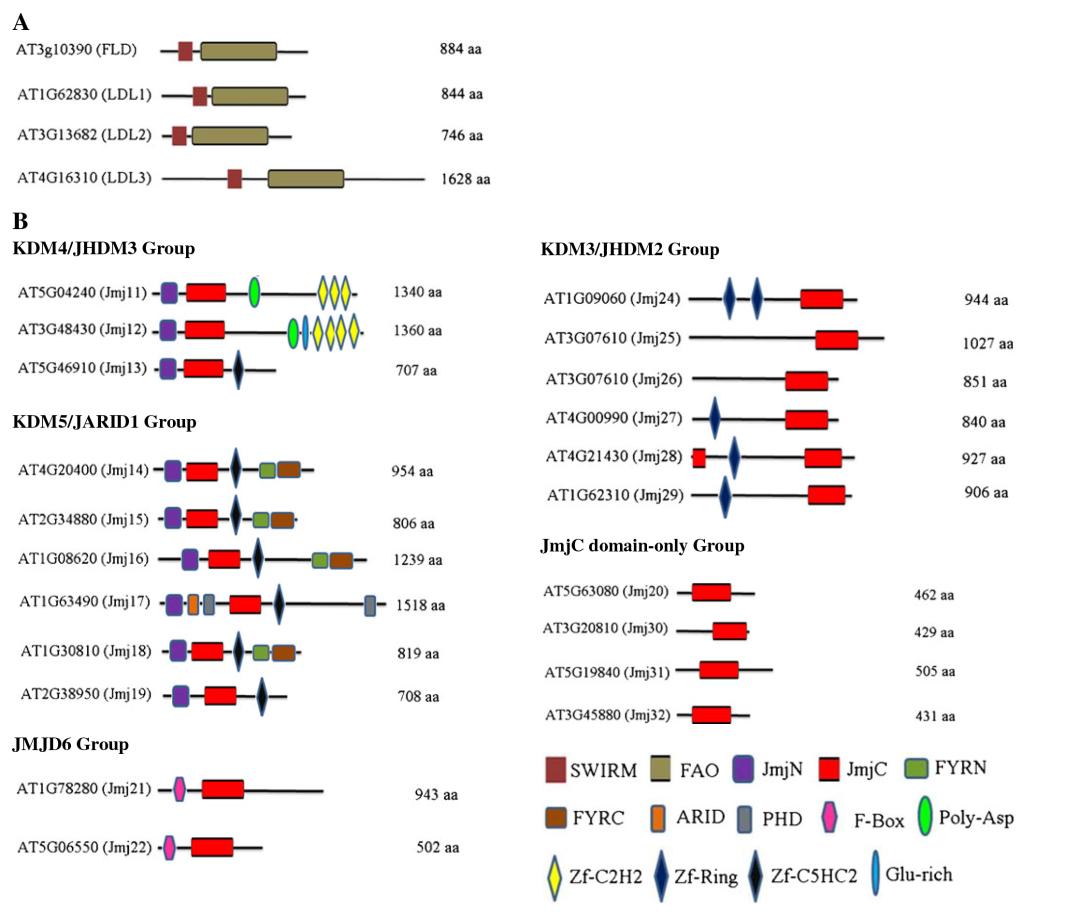 Figure 8 illustrates the conserved domains of Arabidopsis thaliana lysine demethylases (Luo et al., 2013). (A) The conserved structure and domains of KDM1/LSD1 protein. (B) Conserved domains of the Jumonji C (JmjC) domain-containing protein family.
Figure 8 illustrates the conserved domains of Arabidopsis thaliana lysine demethylases (Luo et al., 2013). (A) The conserved structure and domains of KDM1/LSD1 protein. (B) Conserved domains of the Jumonji C (JmjC) domain-containing protein family.
Histone Methylation Reader Proteins
Recognition of methylated histones (Reader) is facilitated by proteins possessing methyl-binding domains (Liu et al., 2010). In Arabidopsis thaliana, several methylated histone reader proteins such as LHP1/TFL2, ORC1, AtING, AL, and WDR5a have been demonstrated to bind methylated histones in vivo or in vitro.
The chromo domain of LHP1/TFL2 is essential for its binding to H3K27me3 at its target loci, and the association of LHP1/TFR2 is necessary for the stable silencing of H3K27me3-marked genes, such as the flowering repressor gene FLC (Mylne et al., 2006). ORC1 constitutes a large subunit of the Origin Recognition Complex (ORC) for DNA replication initiation. Arabidopsis ORC1a and ORC1b possess a PHD finger domain absent in yeast and animal ORC1. Arabidopsis ORC1 can interact with H3K4me3 marks through the PHD finger domain, which is essential for the transcriptional activation of target genes (Sanchez et al., 2009).
It is not yet fully understood how reader proteins translate histone marks into direct downstream functions. Evidence from animal studies suggests that some known reader proteins are subunits of protein complexes with chromatin remodeling or modifying activities (Taverna et al., 2007).
Histone Phosphorylation Modification
The impact of histone phosphorylation modification on chromatin structure and function primarily manifests through two potential mechanisms. Firstly, the negative charge carried by phosphate groups effectively neutralizes the inherent positive charge of histones, thereby weakening the interaction between histones and DNA, resulting in a significant decrease in affinity. Secondly, the phosphorylation modification process provides favorable conditions for the recognition between histones and proteins, further promoting the recruitment of protein complexes and strengthening protein-protein interactions. It is noteworthy that histone phosphorylation modification often occurs at specific amino acid residues, such as serine, threonine, and tyrosine. Particularly, phosphorylation modification of histone H3 exhibits high conservation during evolution, with its major phosphorylation sites including serine 10 and serine 28 (i.e., H3S10p and H3S28p), as well as threonine 3 and threonine 11 (i.e., H3T3p and H3T11p). These phosphorylation modifications play indispensable roles in the regulation of chromatin structure and function.
Histone Ubiquitination Modification
Ubiquitin, a peptide comprised of 76 amino acids (approximately 8.451 kDa), undergoes ubiquitination—a process in which ubiquitin molecules are specifically modified on target proteins by a series of enzymes including activating enzymes, conjugating enzymes, ligases, and deubiquitinating enzymes. The ubiquitination modification of histones functions in altering chromatin conformation, recruiting and activating downstream proteins, and serving as degradation signals for protein degradation. The most extensively studied are the monoubiquitination of histones H2A and H2B. Monoubiquitination of H2A frequently occurs in heterochromatin, associated with gene silencing. Conversely, monoubiquitination of H2B predominantly exists in active euchromatin, associated with transcriptional activation (Lawrence et al., 2016).
Research conducted by Cao et al. demonstrated that histone H2B monoubiquitination mediated by the rice histone ubiquitination enzymes OsHUB1 and OsHUB2 cooperatively regulates the transcriptional control of anther development in conjunction with H3K4me2 (Cao et al., 2015). Additionally, a study by Chen et al. revealed that the E3 ligase HUB2 (AtHUB2) in Arabidopsis thaliana promotes histone H2B monoubiquitination, and overexpression of AtHUB2 enhances drought resistance in transgenic cotton (Chen et al., 2019).
How to Detect Histone Modification
Research on histone modification states and their variants has traditionally relied on specific site-modification antibodies, chromatin immunoprecipitation (ChIP), and DNA sequencing methodologies. However, these techniques face numerous practical challenges, including high technical complexity, substantial costs, and limited efficacy in revealing the intricate and authentic crosstalk between histone modifications.
Although Western blotting (WB) serves as a common antibody-based method for identifying various histones, it is not without limitations. Specifically, WB technology exhibits the following deficiencies:
Inability to detect unknown protein modifications, being restricted to the recognition of known modifications;
Potential bias due to identical modifications across different histones, which may affect the affinity and specificity towards the target modification;
Lack of capability to identify multiple modification types;
Possibility of non-specific interactions among similar histones.
It is noteworthy that studies have indicated that over 25% of commercial antibodies fail to achieve specificity in detection. Among those with demonstrated specificity, more than 20% still fail in chromatin immunoprecipitation assays. These findings underscore the current challenges in histone modification research and highlight the necessity for more precise and efficient methodologies to overcome existing limitations.
Mass Spectrometry for Detecting Histone Modifications
Over several decades of intensive research and continuous development, mass spectrometry (MS) systems have achieved widespread recognition within the academic community for their sensitivity, accuracy, and efficacy in detection. In the domain of histone research, methodologies based on MS have been comprehensively established and refined, enabling precise characterization of histone modification states and detailed analysis of histone variants. This technological framework provides a robust tool and support for advancing histone research.
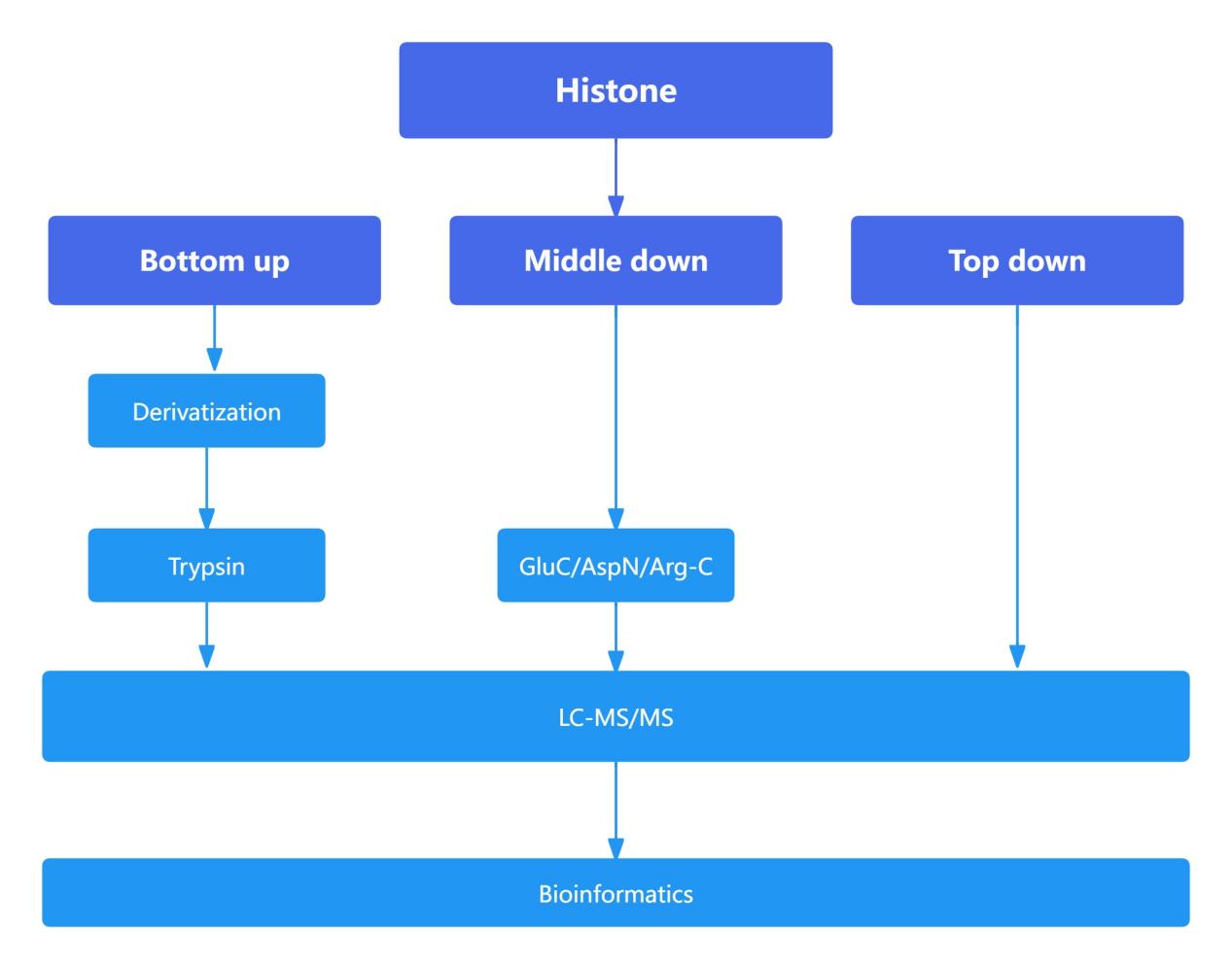 Figure 9. The main process of mass spectrometry system in histone detection
Figure 9. The main process of mass spectrometry system in histone detection
Proteomic Analysis of Histones: Comparison of Bottom-Up, Middle-Down, and Top-Down Approaches
The bottom-up approach is widely employed in proteomics due to its high sensitivity. However, in histone analysis, the high abundance of lysine (Lys) and arginine (Arg) residues, which are cleavage sites for trypsin, leads to excessively small peptide fragments upon enzymatic digestion. This fragmentation complicates subsequent detection and analysis. To enhance detection efficacy and reliability, histones were subjected to derivatization, thereby increasing the length of tryptic peptides.
Compared to the bottom-up approach, which typically generates peptides shorter than 20 amino acids, the middle-down approach yields peptides approximately 50 amino acids in length. These longer peptides facilitate a more detailed analysis of the interplay and influence between histone modification sites that are spatially distant. The top-down approach, which involves the analysis of intact proteins, provides the most comprehensive view of the interrelationships and effects between various modification sites. However, due to the larger size of the fragments analyzed in the middle-down and top-down approaches, these methods exhibit lower sensitivity relative to the bottom-up approach and involve more complex downstream analytical processes.
Research Applications in Histone Modifications
Histone modifications, as the most crucial and diverse regulatory mechanisms in the field of epigenetics, play extensive roles in physiological and pathological processes such as development, organogenesis, tumors, and genetic diseases.
(1) Physiological Studies
Research indicates that enzymes related to histone methylation play pivotal roles in the development of specific organs. These enzymes are indispensable for neurodevelopment and cardiogenesis and also regulate crucial processes such as myogenesis, adipogenesis, and hematopoiesis.
(2) Pathological Investigations
The genesis and progression of cancer cells are not solely attributed to genetic mutations; epigenetic changes also play significant roles.
In-depth analysis of pan-cancer cases reveals that approximately 5% of cancer cases lack identifiable genetic mutations associated with tumorigenesis, suggesting that genetics may not be the sole determinant of cancer. Non-genetic alterations appear to provide an alternative route for the development, progression, and drug resistance of cancer cells.
Studies on pancreatic ductal adenocarcinoma demonstrate that the metastatic process is not driven by driver gene mutations but rather accompanies profound epigenomic reprogramming, further underscoring the pivotal role of epigenetic modifications in cancer development.
Additionally, seminomas are renowned for their extremely low mutation rates, further indicating that cancer arises not solely from DNA mutations but from intertwined genetic and non-genetic processes.
Numerous studies report significant aberrations in histone deacetylases (HDACs) levels in various tumors compared to normal tissues. These alterations are likely to result in changes in histone acetylation status. Delving deeper into the pathological associations between histone modifications and these diseases will aid in the development of novel drug targets capable of maintaining cellular homeostasis and restoring cellular equilibrium.
(3) Drug Targets and Therapeutic Mechanisms Studies
Given the dynamic and reversible nature of histone modifications mediated by writers and erasers, histone modifications and their associated proteins emerge as highly promising drug targets.
(4) Crosstalk Among Multiple Modifications
Histones are often subjected to the combined action of multiple modifications, and these modifications and their associated enzymes collaborate to achieve precise cellular responses. Therefore, in-depth exploration of multiple histone modifications and elucidation of the crosstalk mechanisms between different modifications will provide crucial insights into the dynamic regulation of cellular signals under different conditions.
References
- Bloom, K. and Joglekar, A. 2010. Towards building a chromosome segregation machine. Nature 463, 446–456.
- Cao H, Li X, Wang Z, et al. 2015. Histone H2B Monoubiquitination Mediated by HISTONE MONOUBIQUITINATION1 and HISTONE MONOUBIQUITINATION2 Is Involved in Anther Development by Regulating Tapetum Degradation-Related Genes in Rice. Plant Physiol 168:1389-1405.
- Chen H, Feng H, Zhang X, et al. 2019. An Arabidopsis E3 ligase HUB2 increases histone H2B monoubiquitination and enhances drought tolerance in transgenic cotton. Plant Biotechnol J 17:556-568.
- Creyghton MP, Cheng AW, Welstead GG, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107:21931-21936.
- de la Paz Sanchez M, Gutierrez C. 2009. Arabidopsis ORC1 is a PHD-containing H3K4me3 effector that regulates transcription. Proc Natl Acad Sci U S A 106:2065-2070.
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693-705.
- Lawrence M, Daujat S, Schneider R. 2016. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet 32:42-56.
- Kaya-Okur HS, Wu SJ, Codomo CA, et al. 2019. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10:1930.
- Lee KK, Workman JL. 2007. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol 8:284-295.
- Liu C, Lu F, Cui X, et al. 2010. Histone methylation in higher plants. Annu Rev Plant Biol 61:395-420.
- Luo M, Hung F-Y, Yang S, et al. 2013. Histone Lysine Demethylases and Their Functions in Plants. Plant Molecular Biology Reporter 32:558-565.
- Mylne JS, Barrett L, Tessadori F, Mesnage S, et al. 2006. LHP1, the Arabidopsis homologue of HETEROCHROMATIN PROTEIN1, is required for epigenetic silencing of FLC. Proc Natl Acad Sci U S A 103:5012-5017.
- Taverna SD, Li H, Ruthenburg AJ, et al. 2007. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol 14:1025-1040.
- Ueda M, Seki M. 2020. Histone Modifications Form Epigenetic Regulatory Networks to Regulate Abiotic Stress Response. Plant Physiol 182:15-26.
- Ye Zheng, Kami Ahmad, Steven Henikoff. 2020. CUT&Tag Data Processing and Analysis Tutorial. Protocol.io.
- Zhou H, Liu Y, Liang Y, et al. 2020. The function of histone lysine methylation related SET domain group proteins in plants. Protein Sci 29:1120-1137.
- Kimura H. 2013. Histone modifications for human epigenome analysis. J Hum Genet 58:439-445. doi: 10.1038/jhg.2013.66.
- Torres-Perez JV. 2021. Histone post-translational modifications as potential therapeutic targets for pain management. Trends Pharmacol Sci 42:897-911. doi: 10.1016/j.tips.2021.08.002.
- Zhao S. 2021. The language of chromatin modification in human cancers. Nat Rev Cancer 21:413-430. doi: 10.1038/s41568-021-00357-x.
- Leng X. 2020. AG(enomic)P(ositioning)S(ystem) for Plant RNAPII Transcription. Trends Plant Sci 25:744-764. doi: 10.1016/j.tplants.2020.03.005.
- Kumar V. 2021. Histone acetylation dynamics regulating plant development and stress responses. Cell Mol Life Sci 78:4467-4486. doi: 10.1007/s00018-021-03794-x.
- Wan L. 2017. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543:265-269. doi: 10.1038/nature21687.
- Guo P, et al. 2018. The Histone Acetylation Modifications of Breast Cancer and their Therapeutic Implications. Pathol Oncol Res 24:807-813. doi: 10.1007/s12253-018-0433-5.
- Karch KR, DeNizio JE, et al. 2013. Identification and interrogation of combinatorial histone modifications. Front Genet 4:264.
- Guan X, Rastogi N, et al. 2013. Discovery of Histone Modification Crosstalk Networks by Stable Isotope Labeling of Amino Acids in Cell Culture Mass Spectrometry (SILAC MS). Mol Cell Proteomics 12:2048-2059.
- Moradian A, Kalli A, et al. 2014. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 14:489-497.
- Smith CM, Gafken PR, et al. 2003. Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal Biochem 316:23-33.
Our products and services are for research use only.