What is O-Glycosylation
Protein glycosylation is a vital post-translational modification process observed in the realm of molecular biology. It involves the enzymatic attachment of sugar chains, commonly referred to as glycans, to specific amino acid residues within proteins. Remarkably, it has been estimated that more than half of the proteins within cellular systems undergo glycosylation modifications. This process assumes a pivotal role in orchestrating the structural conformation and functional attributes of proteins, while concurrently maintaining a close nexus with a myriad of physiological processes. The two principal modes of protein glycosylation encompass N-glycosylation and O-glycosylation.
N-glycosylation is characterized by the attachment of glycans to asparagine residues within proteins through the formation of a specific glycosidic bond. Conversely, O-glycosylation pertains to the affixation of glycans to serine or threonine residues within proteins, albeit through a different glycosidic bond. This scholarly work, however, will focus specifically on O-glycosylation, with a specific emphasis on the initiation of mucin-type O-glycosylation, a process catalyzed by the enzymatic action of "peptide: N-acetylgalactosaminyltransferase." This enzyme facilitates the addition of N-acetylgalactosamine (GalNAc) to target proteins.
It is noteworthy that besides GalNAc-initiated O-glycosylation, several other forms of O-glycosylation exist, including O-GlcNAc, O-mannose, O-fucose, O-glucose, and O-xylose. Among these diverse forms, O-GalNAc glycosylation stands out due to its profound biological significance and intricate involvement in various cellular processes.
Mass Spectrometry Analysis of O-Glycosylation
To explore the role of O-glycosylation in diseases or physiological processes, it is necessary to analyze O-glycosylated proteins. This involves identifying which proteins undergo O-glycosylation modifications under different physiological conditions, determining the glycosylation modification sites, elucidating changes in glycan chain structures, and performing quantitative analysis. However, the analysis of O-glycosylated proteins is challenging.
Currently, high-sensitivity, high-throughput mass spectrometry (MS) is a core technology for proteomic analysis and is the preferred method for studying protein glycosylation. O-glycosylation and N-glycosylation both impact the stability, half-life, safety, and biological activity of antibody drugs. Balancing the control of antibody glycosylation modifications within appropriate ranges is crucial. While research related to N-glycosylation is deep and well-established in both academia and industry, thanks to dedicated enzymes and mature sample preparation and analysis methods, O-glycosylation research has rapidly progressed in recent years due to advancements in scientific techniques, as well as the development of relevant reagents and enzymes.
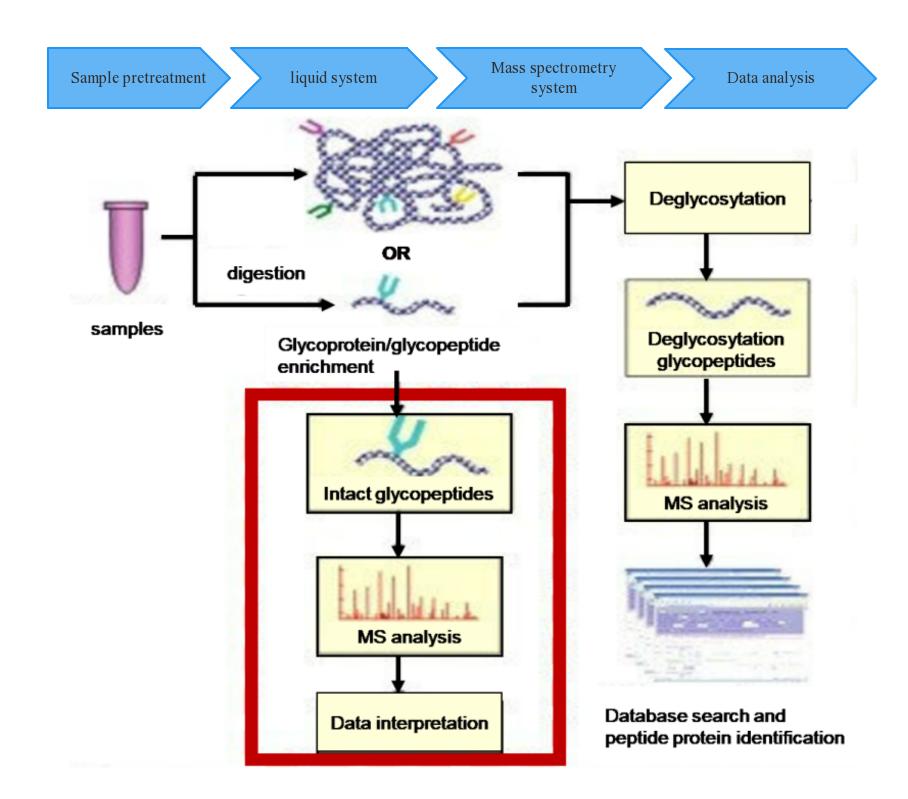
Workflow of O-Glycosylation Mass Spectrometry Analysis
Mass Spectrometry Analysis Service for O-Glycosylation
Creative Proteomics offers customized enzymatic sample digestion solutions tailored to the specific needs of our clients. Our methodology involves the application of proteolytic enzymes for sample treatment, followed by the enzymatic removal of glycan chains attached to peptide segments through the use of glycosidases. Our approach is further complemented by the utilization of our cutting-edge mass spectrometry technology platform, wherein well-established LC-MS/MS protocols are employed for the effective separation and detection of peptide mixtures.
In addition to our experimental procedures, we employ sophisticated data analysis software to rigorously scrutinize the LC-MS and tandem mass spectrometry raw data. This analytical process allows for the confirmation of molecular weight alterations in comparison to the theoretical molecular weight following deglycosylation. Furthermore, our data analysis identifies the specific peptide sequences that undergo glycosylation modifications, ultimately elucidating the precise glycosylation sites and furnishing comprehensive insights into the nature of these glycosylation events.
Sample Requirements
Protein Sample: Quantity >100 μg.
Sample Form: Liquid or dry powder.
Storage: Low temperature (dry ice or ice packs).
Service Advantages
Accurate identification and precise localization of O-glycosylation information.
High-quality data capable of pinpointing O-glycopeptides with up to 7 O-glycosylation sites.
Clear differentiation of positional isomers of O-glycopeptides with complete or nearly complete sequence ion series.
Elucidation of complex glycosylation profiles in protein drug therapy.
Sample Requirements
| Sample Type |
Sample Requirements |
| Plant Leaves |
Wet weight ≥ 3g |
| Plant Roots, Stems, Bark, etc. |
Wet weight ≥ 5g |
| Cell Samples |
Cell count (wet weight) ≥ 5*107 |
| Tissue Samples (Human, Animal, Microbial) |
Wet weight ≥ 50mg |
| Fluid Samples (Serum, Plasma) |
Volume ≥ 200μl |
| Fluid Samples (Cerebrospinal Fluid) |
Volume ≥ 500μl |
| Fluid Samples (Urine) |
Volume ≥ 1000μl |
Characteristics of O-Glycoproteins and Strategies for Mass Spectrometry Analysis
Compared to N-glycoproteins, which are characterized by higher abundance and a single core structure, O-glycoproteins exhibit lower abundance and greater complexity due to multiple core structures. Consequently, the mass spectrometry analysis of O-glycoproteins is inherently more challenging and complex.
One of the primary challenges in O-glycoprotein analysis arises from the limited availability of efficient O-glycosidases and the small size and high hydrophilicity of O-linked glycans. This results in lower sensitivity and difficulty in analyzing O-glycoproteins at the level of O-linked glycans. Currently, O-glycosidases are only available for core structures I and III. For more extended structures, O-linked glycans must first be enzymatically cleaved into core structures using other glycosidases before employing O-glycosidases. Chemical removal methods often alter glycosylation sites and structures to some extent and lack specificity.
Given the very low abundance of O-glycoproteins and the presence of more abundant N-glycoproteins, the analysis of O-glycosylation on O-glycoproteins typically involves the prior removal of N-glycosylation and N-linked sugars using enzymes like PNGase F. Current O-glycosylation analysis primarily focuses on site-specific O-glycoproteomics characterization at the intact O-glycopeptide level. To address the higher competition and suppression from more abundant N-glycopeptides, it is essential to enrich O-glycopeptides from the peptide mixture. Common O-glycosylation enrichment methods include polysaccharide-based chemical or enzymatic oxidation, click chemistry-based metabolic labeling with alkynes or azides, hydrophilic interaction chromatography (HILIC)-based methods, lectin affinity-based methods, among others.
Table 1. HILIC bonded phases, solid supports, and functional groups[4]
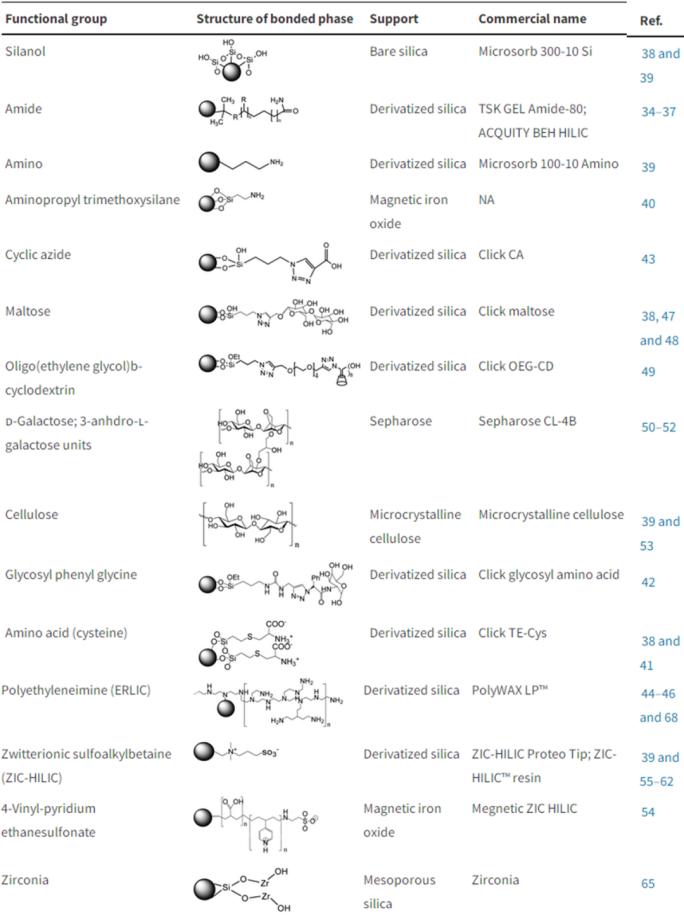
Table 2. Common Lectins for Glycoprotein or Glycopeptide Enrichment[4]
Due to the lack of specific target amino acid motifs for O-glycosylation, and with the target amino acids being Ser and Thr, the precise localization of O-glycosylation sites is challenging. Efficient tandem mass spectrometry dissociation methods are required to generate site-determining fragment ions between two adjacent O-glycosylation sites for accurate site mapping. High-energy collision-induced dissociation (HCD, Figure 1) [5], dissociation methods that include electron transfer dissociation (ETD), electron transfer/higher-energy collision dissociation (EThcD, Figure 2) [6], and photon-based dissociation (such as UVPD) are currently efficient methods that can be employed for this purpose.
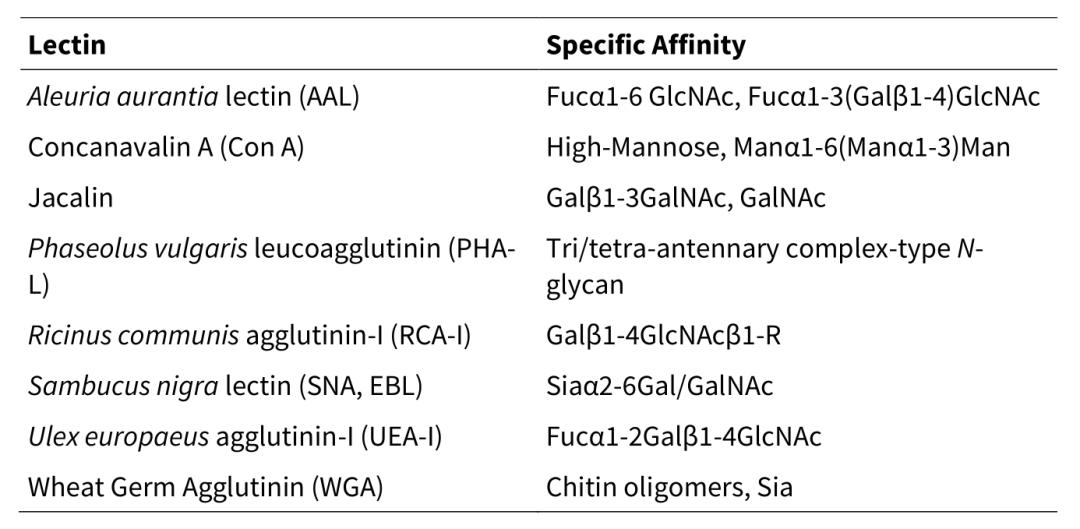
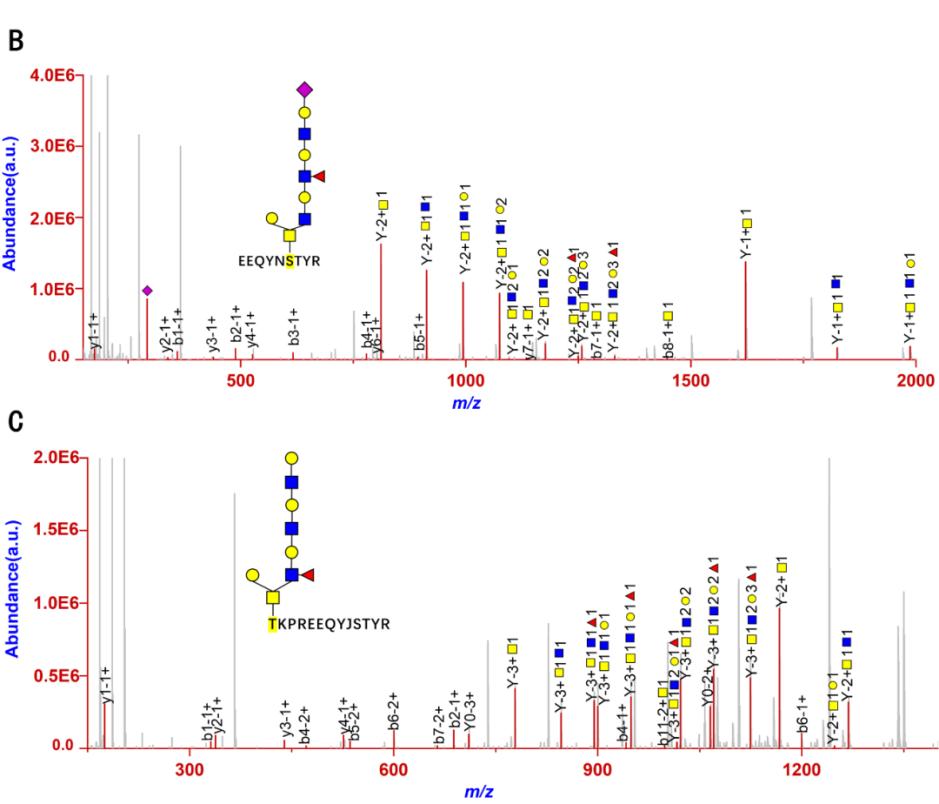 Figure 1. High-energy collision-induced dissociation (HCD) of intact O-glycopeptides IgG1_EEQYNSTYR_N4H4FS1 (B) and IgG1_TKPREEQYNSTYR_N4H4F1S0 (C) in an Orbitrap mass spectrometer [5].
Figure 1. High-energy collision-induced dissociation (HCD) of intact O-glycopeptides IgG1_EEQYNSTYR_N4H4FS1 (B) and IgG1_TKPREEQYNSTYR_N4H4F1S0 (C) in an Orbitrap mass spectrometer [5].
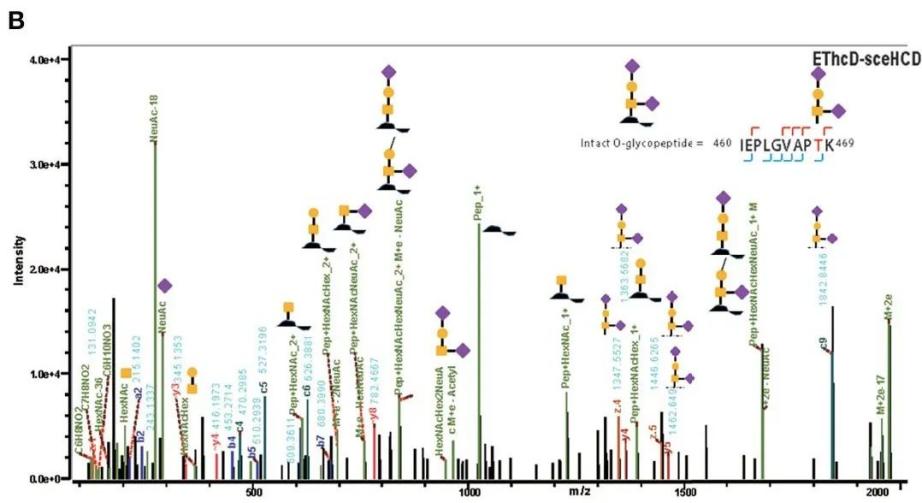
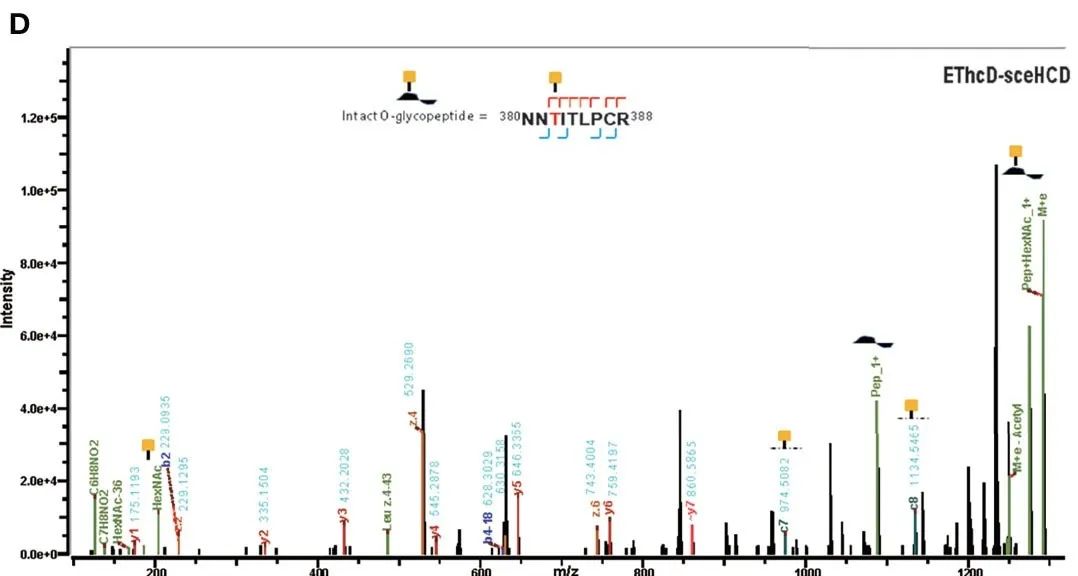 Figure 2. EThcd-sceHCD dissociation spectra of intact O-glycopeptides with one (T468, B) and two (T382, T384) potential O-glycosylation sites[6].
Figure 2. EThcd-sceHCD dissociation spectra of intact O-glycopeptides with one (T468, B) and two (T382, T384) potential O-glycosylation sites[6].
In the context of database searching, identification, and quantification of O-glycosylation at the intact O-glycopeptide level, conventional peptide search engines can be used when interested in a limited number of O-linked glycan modifications. However, when dealing with hundreds or even thousands of O-linked glycans of interest, specialized search engines with theoretical O-linked glycan databases are required.
For site-specific O-glycosylation quantification, conventional peptide labeling quantification methods (e.g., paired isobaric diethyl labeling [7], multiplexed isobaric TMT labeling [5], etc.) can be employed for labeling and quantification of intact O-glycopeptides.
Given the complexity of O-linked glycan structures on O-glycoproteins, the dense modification sites, the relatively small molecular structure compared to N-linked glycans, and the limited tandem mass spectrometry structural features, O-glycoproteomics at both the intact O-glycopeptide and monosaccharide composition levels is currently the preferred method for high-throughput analysis of O-glycosylation. The interactions between multiple O-glycosylations on the same protein and other post-translational modifications need dynamic exploration of these combinatorial modifications, including conventional peptides (e.g., trypsin peptides), larger peptides (e.g., Asp-N and Glu-C peptides), and intact proteins (without enzymatic cleavage) at the single-molecule length and size levels.
References
- Halim, A.; Brinkmalm, G.; Ruetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J., Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. P Natl Acad Sci USA 2011, 108 (29), 11848-11853.
- Steentoft, C.; Vakhrushev, S. Y.; Vester-Christensen, M. B.; Schjoldager, K. T. B. G.; Kong, Y.; Bennett, E. P.; Mandel, U.; Wandall, H.; Levery, S. B.; Clausen, H., Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods 2011, 8 (11), 977-982.
- Levery, S. B.; Steentoft, C.; Halim, A.; Narimatsu, Y.; Clausen, H.; Vakhrushev, S. Y., Advances in mass spectrometry driven O-glycoproteomics. Biochim Biophys Acta 2015, 1850 (1), 33-42.
- Chen, C. C.; Su, W. C.; Huang, B. Y.; Chen, Y. J.; Tai, H. C.; Obena, R. P., Interaction modes and approaches to glycopeptide and glycoprotein enrichment. Analyst 2014, 139 (4), 688-704.
- Zhang, Z.; Xu, H.; Tian, Z., Exploration of quantitative site-specific serum O-glycoproteomics with isobaric labeling for the discovery of putative O-glycoprotein biomarkers. Proteomics Clin Appl 2022, e2100095.
- Zhang, Y.; Zheng, S. S.; Zhao, W. J.; Mao, Y. H.; Cao, W.; Zeng, W. J.; Liu, Y. Q.; Hu, L. Q.; Gong, M.; Cheng, J. Q.; Chen, Y. N.; Yang, H., Sequential Analysis of the N/O-Glycosylation of Heavily Glycosylated HIV-1 gp120 Using EThcD-sceHCD-MS/MS. Front Immunol 2021, 12.
- Wang, Y.; Xiao, K. J.; Tian, Z. X., Quantitative N-glycoproteomics using stable isotopic diethyl labeling. Talanta 2020, 219.
Our products and services are for research use only.