
Principle and Advantages of SWATH-MS Technology
Online InquirySWATH can be applied to proteomics, metabolic lipidomics, pharmaceutical and biopharmaceutical, food screening, environmental analysis and toxicology analysis. In proteomics research, the high-throughput quantitative capability of SWATH batches to detect thousands of proteins and hundreds of thousands of peptides has been validated in a large body of literature. How to achieve reproducibility of data results within and across laboratories has been a difficult problem in the industry, and SWATH technology is considered to be a very good solution to this challenge.
Working Mode of SWATH
SWATH-MS detection typically relies on a first-stage fast scan of the mass spectrum (typically at least 10 Hz), high resolution (typically at least 15,000 FWHM resolution for MS2), and high mass accuracy (at least 50 ppm) with a common configuration of a first-stage mass analyzer with a quadrupole (Q) and a second-stage mass analyzer with a time-of-flight (TOF) or Orbitrap.
The mass spectrum is scanned according to the job tasks set in the method. In the conventional setup of SWATH, within a scan cycle, the first step is to command the mass spectrometer to acquire a first level full scan spectrum; the second step is to partition the parent ions in the scan range by a certain mass range. For example, if the scan range is 50-1000 Da, and the computer sets the Q1 partition window to 50 Da, then it is divided into 50-100 Da, 99-150 Da, 149-200 Da in order .... Q1 will send these specific mass ranges, in turn, to Q2 to break them up, and then obtain all the fragmentation information of the parent ion in the corresponding mass interval. Of course, the Q1 window can be set to other values or a variable window can be set.
To ensure sufficiently fast recording of MS1 and MS2 for peak extraction and peptide quantification, optimal trade-offs need to be made in terms of the number of MS2 scans, the accumulation time of each scan, the size of the parent ion isolation window, the peptide scan mass range, and other parameters.
SWATH uses a data independent acquisition mode. The acquired information is processed by the software to obtain primary quantitative and qualitative information for each peak, secondary qualitative information, and MRM-like secondary quantitative information. Thus, SWATH is a truly panoramic, high-throughput, data-traceable mass spectrometry technique.
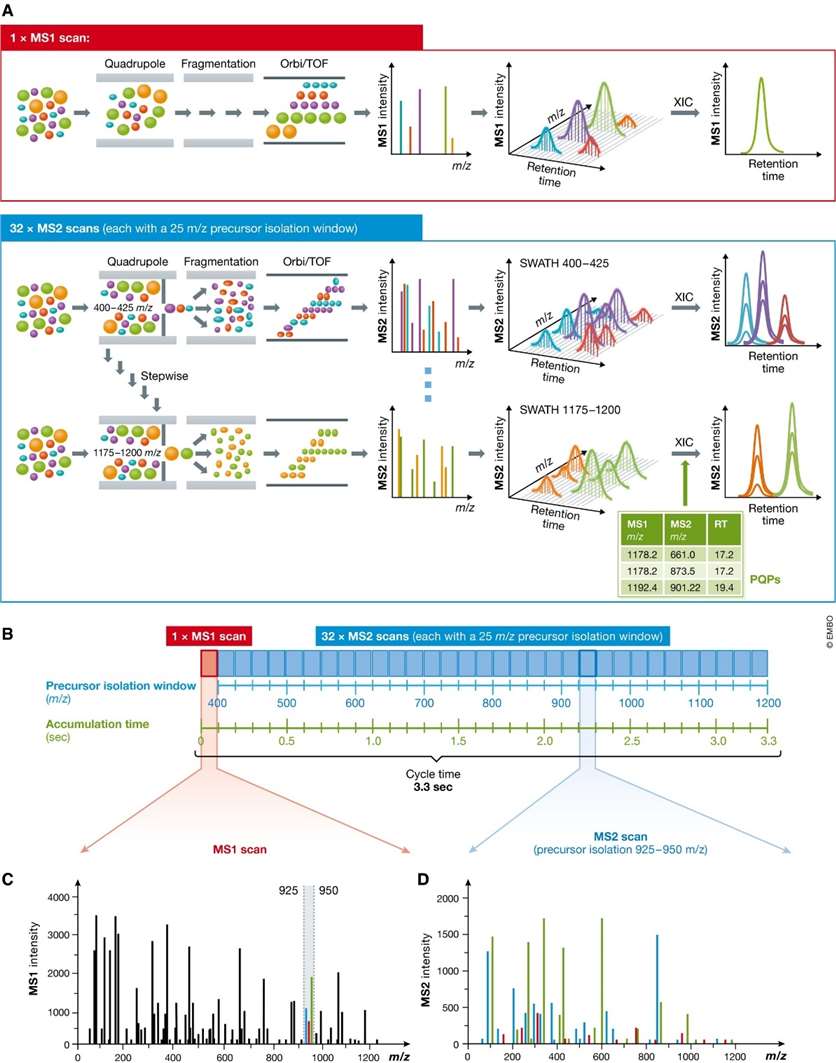 Principle of sequentially windowed data-independent acquisition in SWATH-MS (Ludwig et
al., 2018)
Principle of sequentially windowed data-independent acquisition in SWATH-MS (Ludwig et
al., 2018)
Advantages of SWATH-MS
SWATH-MS (Sequential Window Acquisition of All Theoretical Fragment Ion spectra) has several advantages, including:
Comprehensive analysis: SWATH-MS allows for comprehensive analysis of large and complex proteomes, providing detailed information about protein expression levels and post-translational modifications.
High Sensitivity: The technique is highly sensitive and can detect even low abundance proteins and modifications.
Multiplexing capability: SWATH-MS enables multiplexed analysis, enabling the simultaneous analysis of multiple samples.
Reduced sample preparation: Unlike other mass spectrometry-based methods, SWATH-MS requires minimal sample preparation and allows for the direct analysis of complex biological samples.
Quantitative analysis: SWATH-MS provides quantitative information about protein expression levels and post-translational modifications, making it useful for biomarker discovery and functional studies.
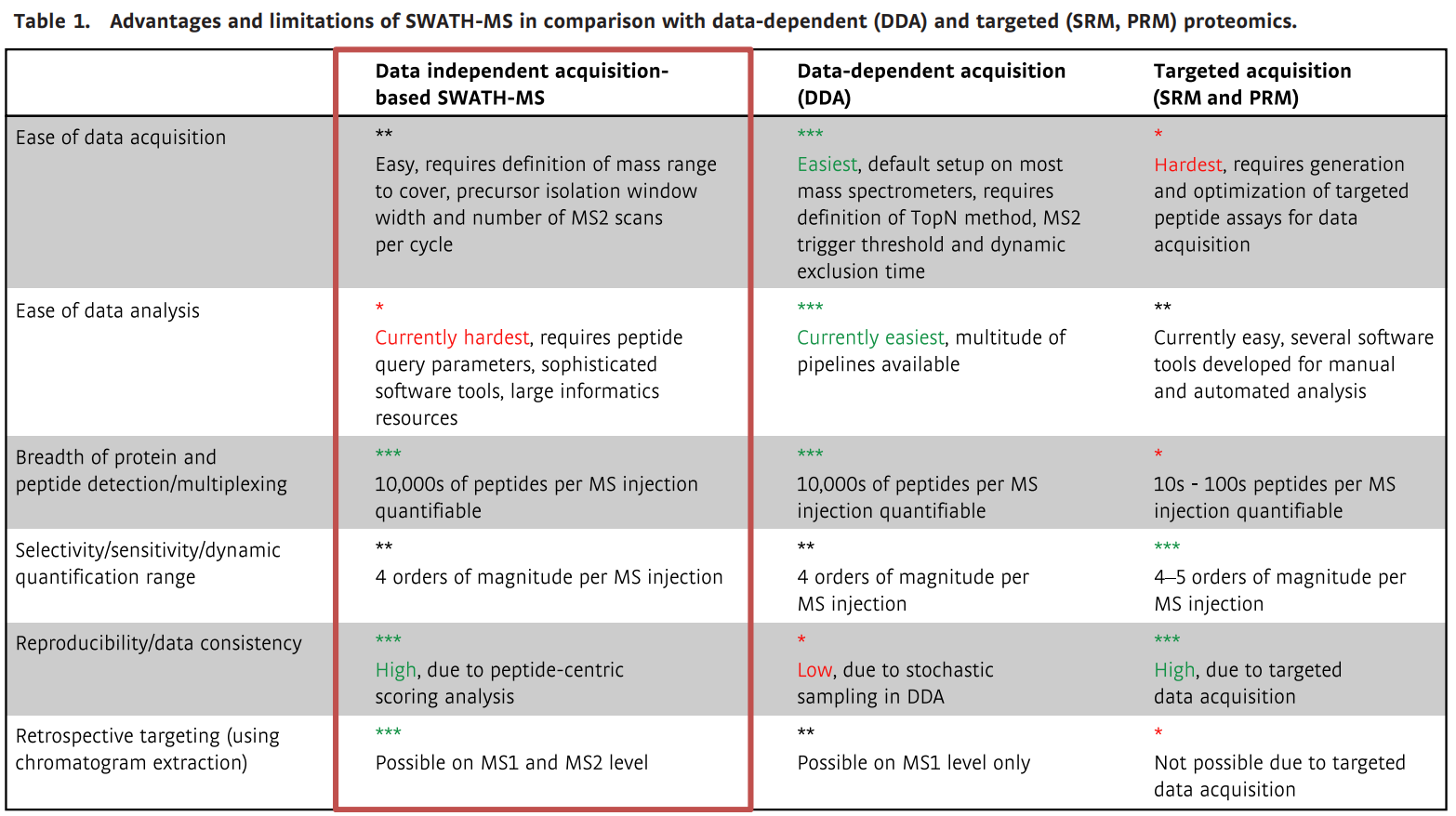 Advantages and limitations of SWATH-MS in comparison with data-dependent (DDA) and
targeted (SRM, PRM) proteomics (Ludwig et al., 2018)
Advantages and limitations of SWATH-MS in comparison with data-dependent (DDA) and
targeted (SRM, PRM) proteomics (Ludwig et al., 2018)
An important technical advantage of SWATH-MS is the simplified data acquisition process. One SWATH-MS acquisition requires the definition of the parent ion isolation method (including window peak width, m/z acquisition range, and MS2 scan accumulation time). Once the method is confirmed, all similar types of samples in the same system can now be analyzed experimentally using the same method in a uniform manner. Even if the sample types differ, the same acquisition method has proven to be still reliable.
SWATH-MS achieves a huge increase in the number of quantitative peptides. The more peptides quantified, the more proteins can be quantified, and the higher the coverage of protein sequences detected. Over 10,000 peptides can be characterized in a single SWATH-MS assay.
When studying the proteomics of hundreds of samples, the reproducibility of peptide detection and consistency of quantification becomes the main difficulty. It is very difficult to require a high degree of consistency in peptide quantification in DDA proteomic experiments, because you cannot require that the heuristic parent ion selection approach used for real-time signal acquisition by mass spectrometry DDA be reproducible. So when a peptide is not successfully characterized in a particular DDA assay, you cannot tell whether the peptide is detectable or not ("true negative peptide quantification"). The actual situation is probably just because the mass spectrometry happened to not fragment the peptide because of the presence of a bunch of co-flux peptides ("false negative peptide quantification"). SWATH-MS can solve the above problem.
The information on the MS1 parent ion and MS2 fragment ion peaks of both chromatography and mass spectrometry of the SWATH-MS data is quite complete, which also facilitates retrospective analysis after a period of time.
Reference
- Ludwig, Christina, et al. "Data‐independent acquisition‐based SWATH‐MS for quantitative proteomics: a tutorial." Molecular systems biology 14.8 (2018): e8126.
Related Services
* For Research Use Only. Not for use in diagnostic procedures.
