
iTRAQ/TMT, Label Free, DIA, DDA in Proteomic
Online InquiryDifference between iTRAQ/TMT and Label Free
Isobaric tags for relative and absolute quantitation (iTRAQ) and tandem mass tag (TMT) are the two most widely used in vitro peptide labeling and quantification technologies developed by Thermo and AB SCIEX, respectively, with similar principles. The main difference is that TMT can simultaneously label up to 16 different biological samples, while iTRAQ can simultaneously label up to 8 different biological samples.
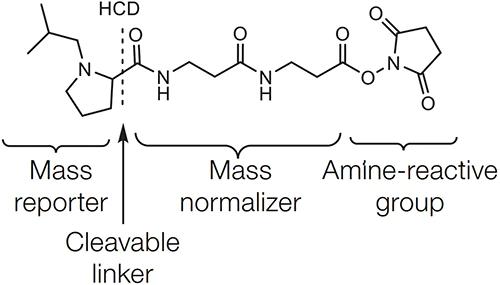 Diagram of the chemical structure of the TMT label
Diagram of the chemical structure of the TMT label
Let's take TMT as an example to illustrate the label structure. All labels in a set of TMT labeling reagents carry the same mass. The TMT chemical structure consists of a mass reporting group, a mass balance group, and a peptide reaction group. The peptide reactive group can covalently bind to the free amino group of the N-term or terminal lysine side chain group of the peptide, thus labeling the entire peptide with a TMT tag. Different TMT tags have different masses of the reporter group, and the total molecular mass of the different TMT tags is kept the same by the balance of mass balance groups. As the peptide enters the secondary mass spectrum, the mass reporting group is released due to fragmentation, producing a reporter ion peak in the low mass region of the mass spectrum. The intensity of this mass spectrometric signal is a response to the relative expression of a particular peptide in different samples.
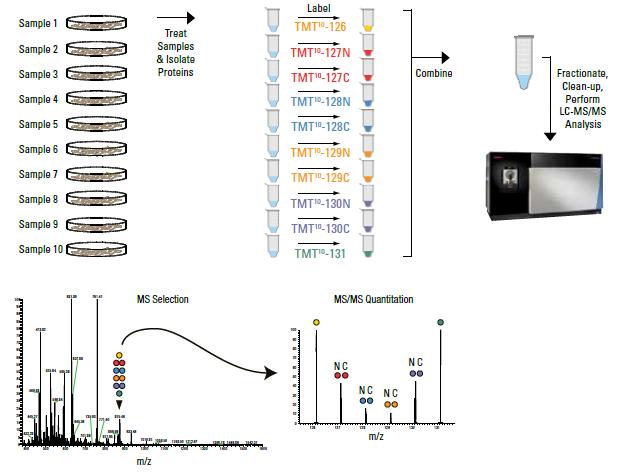
The use of isotopes allows the labeling of different samples. The same batch of samples is then tested together on the machine. The expression of different proteins in each sample can be distinguished according to the different isotopic labeling.
Advantages of TMT: accurate quantification, good data reproducibility, high sensitivity low sample requirement and high number of detected proteins.
Disadvantages of TMT: Since there are only up to 16 isotope markers, the large sample volume requires the addition of internal reference samples at each loading, which increases the cost. In addition, it is not possible to reject outlier samples for reanalysis.
In contrast to TMT for protein labeling and quantification, label free eliminates the need for expensive stable isotope labels as internal standards and each sample is individually loaded. It is only necessary to analyze the mass spectrometry data generated during the large-scale identification of proteins and compare the signal intensity of the corresponding peptide in different samples for the relative quantification of the peptide corresponding to the protein.
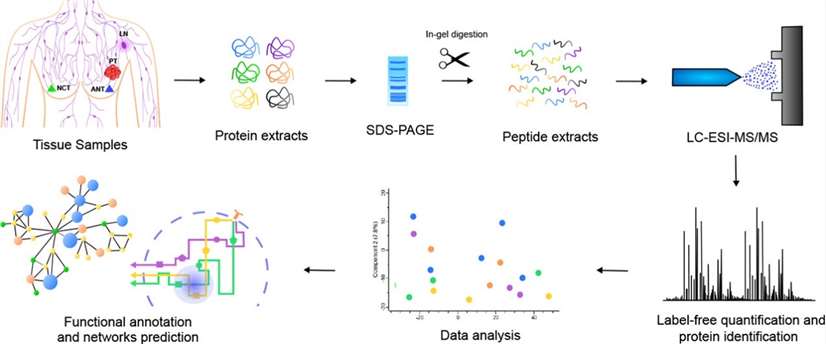 Quantitative label-free mass spectrometry (Gomig et al., 2019)
Quantitative label-free mass spectrometry (Gomig et al., 2019)
Advantages of label free: no reliance on isotope labeling, individual detection for each case. Therefore, the experimental design is flexible and suitable for clinical testing of tens or hundreds of samples. It is also possible to observe the "presence or absence" of proteins in the sample.
Disadvantages of label free: The stability of the instrument and the operation of the laboratory personnel are very important factors that may produce systematic errors.
Differences between DIA and DDA
The routine process of LC-MS/MS protein profiling: sample preparation, extraction and purification of proteins, trypsin digestion into peptides, liquid chromatography separation (LC), primary mass spectrometry detection (parent ion signal), peptide fragmentation into secondary mass spectrometry detection (for determining amino acid sequences), and data analysis.
Data independent acquisition (DIA) and data dependent acquisition (DDA) refer to two modes of data acquisition for performing secondary mass spectrometry. DDA is a selective data acquisition mode, while DIA is a full scan data acquisition mode.
| Data Dependent Acquisition | Data Independent Acquisition (DIA) | |
|---|---|---|
| Description | A specific number of peptide molecules (e.g. TOP20 with the strongest signal intensity) in the -level mass spectra are randomly selected for entry into the secondary mass spectra for fragmentation. | Full scan. Secondary mass spectrometry fragmentation for all ions in each window. |
| Features | The mass spectrometry data is less complex and relatively less difficult to analyze. However, random selection always favors high-abundance peptides. Therefore, low-abundance peptides are missing more. This defect is further amplified when large sample size analysis is performed, presenting a higher number of missing values. | Complex data for mass spectrometry results and high requirements for data analysis. Comprehensive data coverage, no bias. |
| Applications | Small-scale samples | More suitable for protein detection of large-scale samples and complex systems. |
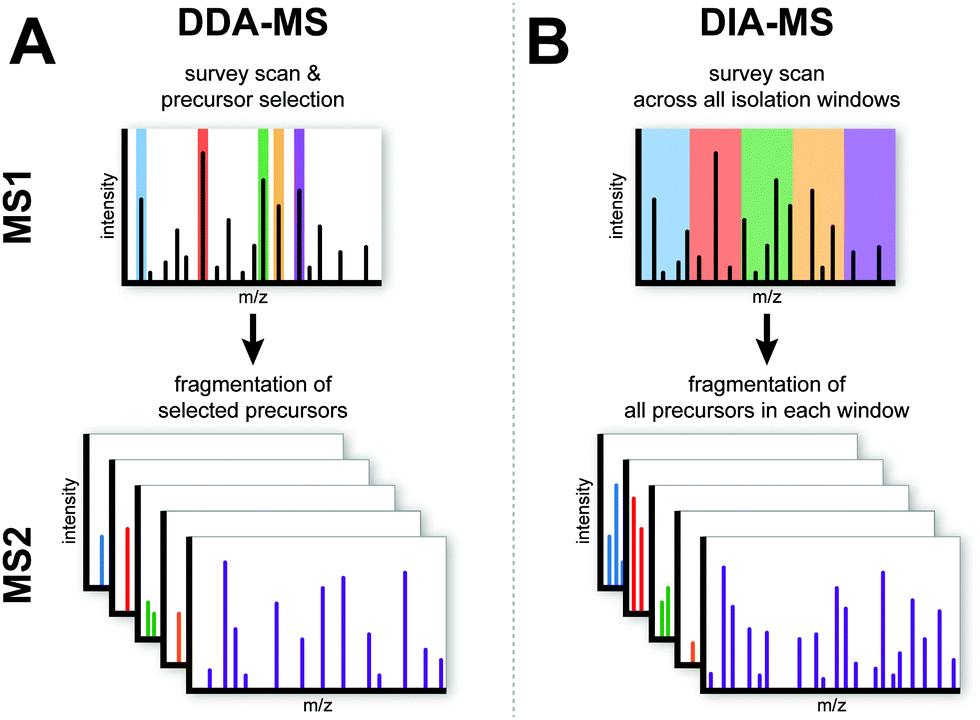 Schematic overview of the DDA-MS and DIA-MS (Krasny et al., 2021)
Schematic overview of the DDA-MS and DIA-MS (Krasny et al., 2021)
References
- Gomig, T. H. B., Cavalli, I. J., et al. (2019). Quantitative label-free mass spectrometry using contralateral and adjacent breast tissues reveal differentially expressed proteins and their predicted impacts on pathways and cellular functions in breast cancer. Journal of proteomics, 199, 1-14.
- Krasny, L., & Huang, P. H. (2021). Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Molecular omics, 17(1), 29-42.
* For Research Use Only. Not for use in diagnostic procedures.
