DIA Mass Spectrometry: A New Tool For High-Throughput Phosphoproteomics
Protein phosphorylation, an essential protein post-translational modification, has the potential to exert a significant impact on protein function by modifying its activity, subcellular localization, interaction, and stability. Moreover, owing to its regulatory influence on signaling pathways implicated in severe ailments, such as cancer, the research surrounding phosphorylation, including its modification sites, has been a topic of great interest and a perennial challenge for scientists.
A Innovative Phosphoproteomics Workflow
Nature Communications (J. Olsen et al., 2020) published an astonishingly rapid and innovative phosphoproteomics workflow rooted in data independent acquisition (DIA) mass spectrometry. This groundbreaking methodology grants control over the acquisition time of single-shot data, remarkably within a mere 15 minutes, and furnishes the scientific community with a novel tool for phosphoproteomics.
In existing workflows for phosphoproteomics, in order to achieve the highest possible analysis depth, in addition to enriching for phosphorylated peptides, there are also cumbersome sample fractionation steps that typically take several days or even a week, limiting its clinical application. In order to achieve high-throughput, high-accuracy phosphoproteomics analysis, the Olsen group first optimized the DIA data acquisition method based on the existing DIA collection method with a 15-minute liquid chromatography separation time as the baseline. Based on the characteristics of the Thermo QE HF-X instrument used in the experiment, the group optimized parameters such as collision energy, adjacent window width (overlap of DIA windows), scan cycle, and HCD resolution to obtain optimal mass spectrometry acquisition parameters. Using a test sample of starting peptides of 200 μg, DDA and DIA data acquisition methods were tested, and in the data analysis process, directDIA (annotation 1) and traditional library-based DIA methods were also used for analysis (Figure 1a) for DIA data. As can be seen from the results, the DIA method outperforms the DDA method in terms of the number of identified phosphopeptides (nearly 2 times more phosphopeptides with phosphorylation sites, nearly 28,000 phosphopeptides) and reproducibility (R2 0.93 vs 0.89), especially noteworthy is that the dynamic range of the DIA method is an order of magnitude higher than that of the DDA method (Figures 1e-f), which the authors speculate is due to the higher ion utilization efficiency of the DIA method, using the MS2 target value setting in its instrument parameters (Figures 1g-h). In the context of data analysis, directDIA refers to comparing deconvoluted DIA data with protein sequence databases, instead of using the commonly used spectral libraries in DIA analysis.
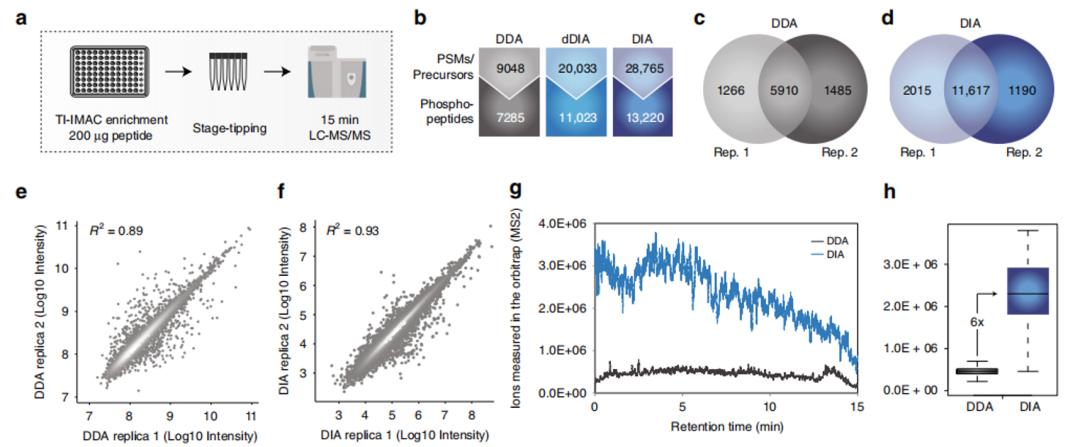 Figure 1 |a-h Comparison of experimental results between DDA and DIA
Figure 1 |a-h Comparison of experimental results between DDA and DIA
To further compare the performance of DIA and DDA in terms of quantitative accuracy, the authors spiked yeast phosphorylated peptides at predetermined ratios into Hela peptides (as background), and performed data acquisition using the same experimental workflow. In the data analysis process, MaxQuant software (MQ, for DDA analysis) and Spectronaut software (SQ, for DIA analysis) were used for analysis, and the results were processed using scripts for method comparison (Figure 2.I). From the measured ratios (Figure 2.J), error rates (Figure 2.K), and ROC curves (Figure 2.I), it can be seen that the performance of DIA method is also superior to that of DDA method, especially when the sample concentration is high (2:1), which is of great significance for clinical samples.
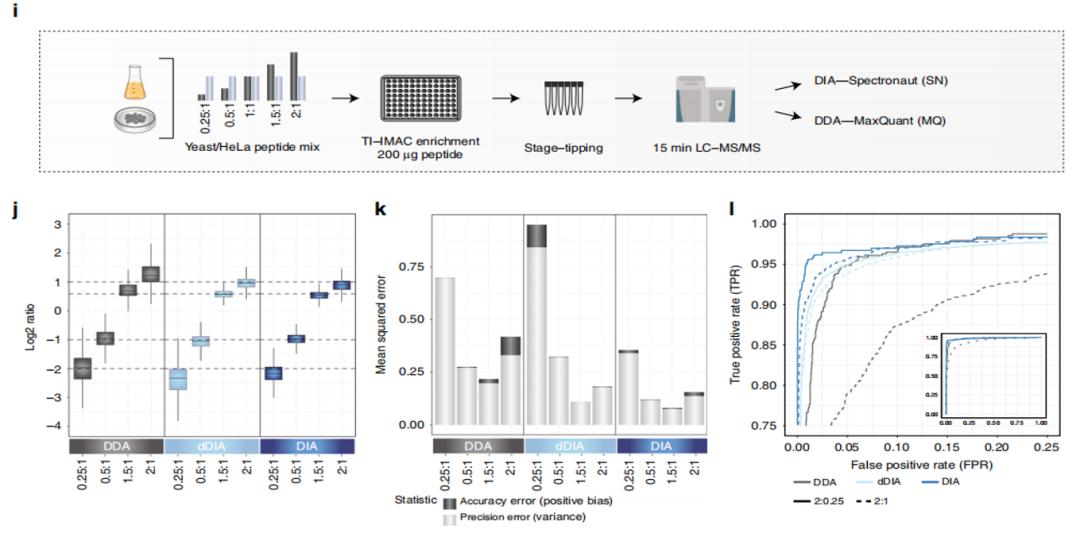 Figure 2 | i-l Comparison of DDA and DIA experimental results.
Figure 2 | i-l Comparison of DDA and DIA experimental results.
The analysis of phosphorylation data also has an additional difficulty, which is the localization of phosphorylation sites. To address the characteristics of the DIA acquisition method (wide isolation window for precursor ions), the authors developed a PTM localization algorithm to accurately locate phosphorylation sites. In simple terms, this algorithm first detects and classifies all peak groups, and then uses an enumeration method to assign them to possible sites. By combining other information, the algorithm divides the fragment ions into confirming or refuting ions for the site. Finally, it calculates the score of all confirming ions and subtracts the score of refuting ions to obtain the final score (Figure 3). To validate the algorithm, the authors analyzed a synthetic phosphorylated peptide with known site(s) using the optimized experimental workflow (Figure 3). The results showed that the DIA method had lower error rates and higher accuracy. Currently, this algorithm has been integrated into the Spectronaut software, which is compatible with DIA workflows.
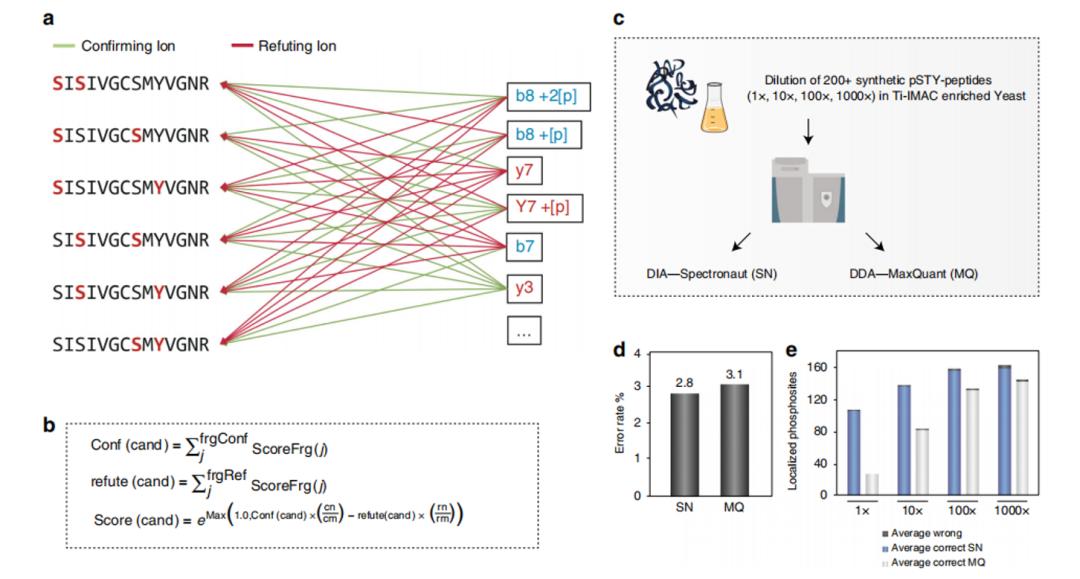 Figure 3 | PTM Localization Algorithm Example and Experimental Results.
Figure 3 | PTM Localization Algorithm Example and Experimental Results.
The authors then applied this workflow to study cell signaling pathways in retinal pigment epithelium cells (RPE1) by inducing corresponding signaling pathways using different MEK kinase inhibitors. Data collected by both DDA and DIA methods were analyzed using the MQ and SQ software, respectively. In addition, different analysis methods were applied in the DIA data analysis process, including project-specific library, community-based library, and combined library, in addition to the directDIA method (Figure 4). The overall results showed that DIA performed better than DDA (except for directDIA, which was similar to DDA) (Figure 4b). The heat map (Figure 4c) and motif enrichment analysis (Figure 4e) also showed that the phosphoprotein/position patterns identified by DDA and DIA were similar, and the enriched positions were consistent with the signaling pathway, confirming the reliability of the method. The authors then extended this method to stoichiometry analysis of phosphorylation sites and larger-scale kinase inhibition experiments, which validated the reliability and scalability of this method.
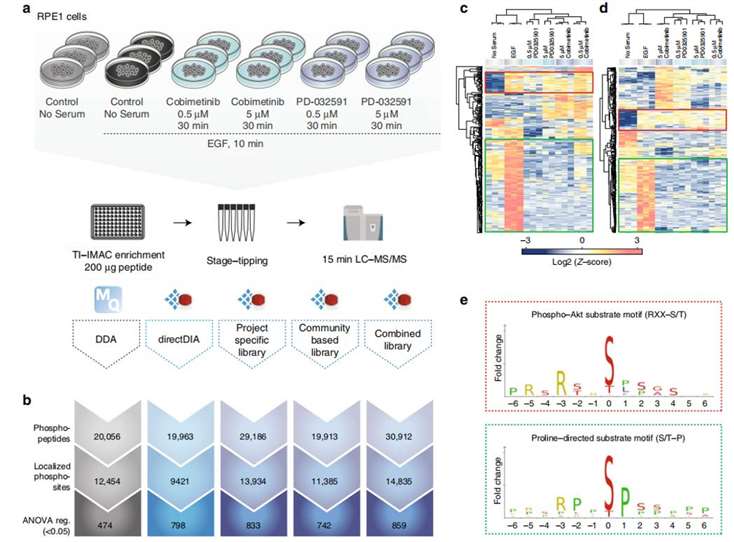 Figure4 | Validation experiments of signaling pathways
Figure4 | Validation experiments of signaling pathways
DIA Superior to DDA
In the final discussion, the authors pointed out that their method, including the mass spectrometry acquisition parameters and the PTM localization algorithm, has been integrated into the DIA data analysis workflow, and its performance has been validated to be superior to the DDA method. Moreover, due to the sensitivity advantage of the DIA method, the sample starting amount in this workflow can be lower than 200 µg, which is more suitable for clinical research needs.
Reference:
- D. B. Bekker-Jensen et al., Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nature Communications 11, (2020).
* For Research Use Only. Not for use in the treatment or diagnosis of disease.

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)