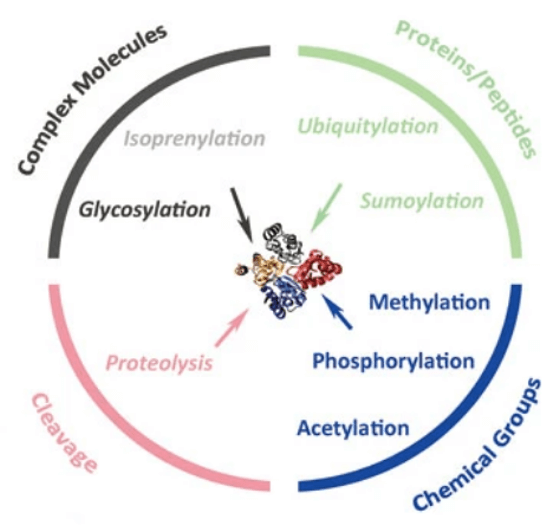
Proteins and peptides are linear polymers made up of combinations of the 20 most common amino acids linked with each other by peptide bonds. The identification and characterization of proteins and peptides is a key step in proteomics. The protein produced by the ribosome may undergo covalent modifications, or post-translational modifications, after its incorporation of amino acids. Over 200 such modifications have been detected already, the most important being glycosylation, the formation of disulfide bridges, phosphorylation, sulfation, hydroxylation, carboxylation and acetylation of the N-terminal acid.
Protein post-translational modifications (PTMS) are chemical modifications of proteins that regulate important biological processes, such as protein signaling, localization and degradation, by affecting protein structure and are involved in the development of a variety of diseases. Therefore, the identification of PTMs at the proteomic level is essential for gaining insight into the role of proteins in different biological processes. However, today, despite their significance, the comprehensive identification and discovery of PTMs in complex biological samples still pose difficult challenges for proteomics techniques, including identifying, site localizing and quantifying these modifications under different conditions (eg. healthy and diseased organisms).
In order to solve the above problems, Creative Proteomics has established unique discovery proteomics to analyze the protein modification of different samples.
 Our PTM Identificaion Service[2]
Our PTM Identificaion Service[2]
Our Post-translational Modifications (PTMs) Analysis Service
Using our advanced Discovery Proteomics Platform, we can quantitatively distinguish different PTM sites, such as phosphorylation, glycosylation, acetylation, methylation, ubiquitination and oxidation, of the same complex peptide on a large scale from biological samples (such as cultures, tissues, or cells in plasma).
We can analyze up to 9,000 proteins per sample under different conditions, and identify and localize protein PTM sites with or without enrichment approach prior to the mass spec analysis. Depending on the complexity of the sample, PTM enrichment may or may not be required during sample preparation. Our Discovery Proteomics Platform is ideal for detecting differences in protein abundance and low-abundance proteins in multiple samples. We also offer highly multiplexed targeted proteomics with absolute quantification for customized panels of proteins.
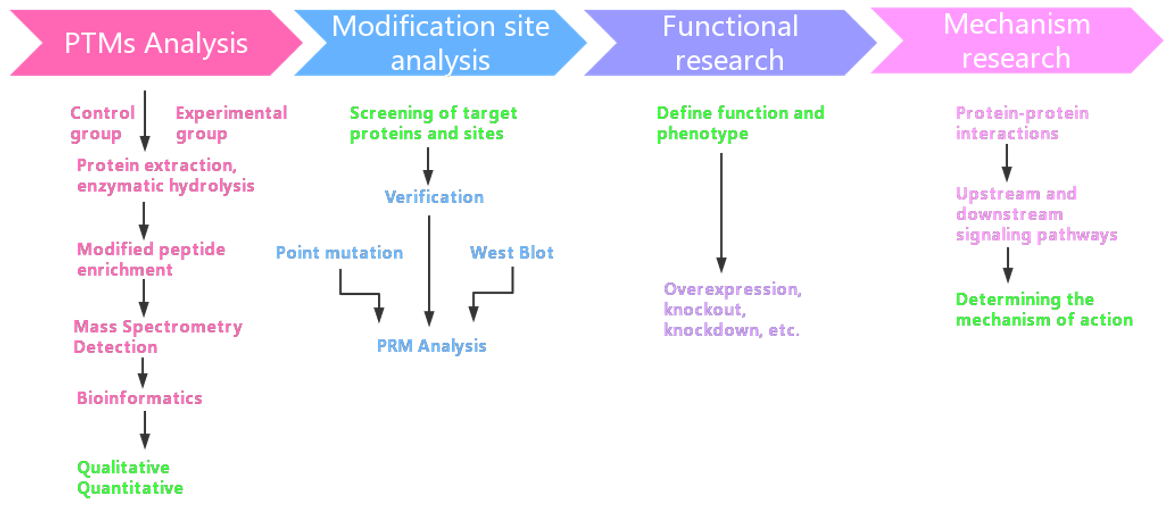
Workflow of PTM Analysis
Sample Preparation: Collect biological samples, such as cell extracts, tissue specimens, or bodily fluids. Perform protein extraction and purification to ensure high-quality protein samples.
Protein Digestion: Digest proteins into peptides using an appropriate protease (e.g., trypsin). Ensure thorough enzymatic digestion to generate peptides suitable for mass spectrometry analysis.
Ionization and Precursor Ion Selection: Employ mass spectrometry for ionization, typically using a high-resolution and high-sensitivity instrument. Perform precursor ion selection using high-energy ionization to ionize peptides.
Data Acquisition: Utilize DIA methodology to divide the entire mass spectrum into windows, collecting data for all fragment ions within each window. Cycle through windows to obtain comprehensive ion spectra data, ensuring the detection of all peptides in highly complex mixtures.
Database Search and PTMs Identification: Employ bioinformatics tools to match experimental data against protein databases. Identify potential PTM sites, such as phosphorylation, methylation, acetylation, etc.
Quantitative Analysis: Quantify identified peptides based on the intensity of mass spectrometric signals, comparing abundance differences between different samples. Use relative or absolute quantification methods depending on the experimental design and objectives.
Statistical Analysis and Biological Interpretation: Conduct statistical analysis on quantitative results to determine significant changes. Combine findings with biological background knowledge to interpret the relevance of identified PTMs to specific biological processes or disease states.
Reporting and Data Sharing: Generate a detailed analysis report, providing information on identified modifications, quantitative results, and relevant statistical information. Share data with clients to support further experimental design or research directions.
Advantages of DIA PTM Analysis
- High accuracy
- High throughput, more than 9000 proteins can be identified at once
- Complete and comprehensive bioinformatics analysis
- High repetition rate
- Competitive price
Sample Requirements of DIA PTM Analysis
Based on a variety of protein extraction technologies, we can quickly extract proteins from various samples, and design personalized experimental programs according to different experiment purposes.
| Sample Type |
Protein |
# of Cells |
Animal Tissue |
Plant Tissue |
Blood |
Urine |
Serum |
Microbes |
| Quantify |
100 ug |
1×107 cells |
1 g |
200 mg |
1 mL |
2 mL |
0.2-0.5 mL |
Dry weighed: 200 mg |
Delivery:
LC-MS/MS Report
References:
- Fanni Bugyi, Dániel Szabó, Győző Szabó, Ágnes Révész, Veronika F. S. Pape, Eszter Soltész-Katona, Eszter Tóth, Orsolya Kovács, Tamás Langó, Károly Vékey, and László Drahos.Influence of Post-Translational Modifications on Protein Identification in Database Searches. ACS Omega 6 11, 7469–7477 (2021).
- Yu-Chieh Wang.Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Research 24, pages 143–160 (2014).
Case Quantitative Profiling of Protein Post-Translational Modifications in NASH Using a 4 Da Precursor Mass Window DIA Method
Background
Protein post-translational modifications (PTMs) play a crucial role in cellular processes, and their dysregulation is implicated in diseases such as non-alcoholic steatohepatitis (NASH). To understand the intricate PTM landscape in NASH, a robust and accurate analytical method is essential.
Samples
The study utilized complex liver lysate samples from mouse models with genetically manipulated S-adenosylmethionine (SAMe) concentrations, specifically Mat1a −/− and Gnmt −/− models, representing different NASH conditions.
Technical Methodology
The methodology employed a 4 Da precursor mass window in a data-independent acquisition (DIA) approach. This method allowed for the differentiation of modified peptidoforms from unmodified peptides, particularly focusing on protein methylation, acetylation, and succinylation. The Thesaurus tool was updated to aid in PTM site localization, offering a graphical interface for end-users.
The study compared multiplexed 4 and 6 Da precursor mass windows with nonmultiplexed windows on complex liver lysate, demonstrating the superior performance of the 4 Da method. Linear protein quantitation was achieved in both 60 and 120 min liquid chromatography (LC) gradients. The methodology addressed challenges associated with large precursor mass window DIA methods, providing a more accurate quantitation of low-abundance peptides and avoiding ambiguity in identification.
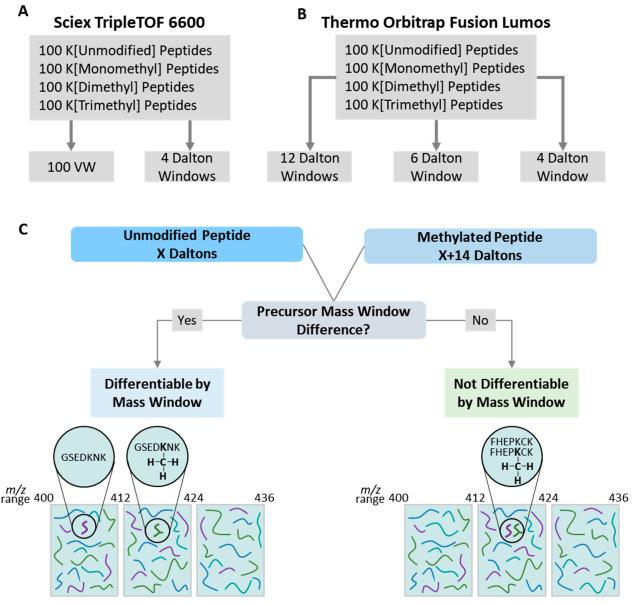 Schematic overview of experimental design.
Schematic overview of experimental design.
Results
The 4 Da precursor mass window DIA method exhibited efficacy in quantifying protein methylation, acetylation, and succinylation, along with total protein quantification, from the same sample and acquisition run. The study successfully applied the approach to the NASH models, revealing insights into differential methylation potential, altered acetylation, and total protein changes. The proposed hypothesis linking PTMs to regulatory functions in mRNA stability, protein synthesis, and mitochondrial function provided a comprehensive understanding of their roles in NASH.
This advanced analytical method opens avenues for studying PTMs in various biological contexts, particularly in diseases characterized by intricate molecular modifications.
Reference:
- Robinson, Aaron E., et al. "Lysine and arginine protein post-translational modifications by enhanced DIA libraries: quantification in murine liver disease." Journal of proteome research 19.10 (2020): 4163-4178.

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)