Global Proteomic Quantification Service
The label-free method is a good alternative for protein quantification because of the higher mass accuracy and faster scanning speed on mass spectrometers, the significant advances in computational methods and software for data processing, and its lower cost compared to labeled methods. For a comprehensive characterization of the proteome, an analytical platform capable of quantifying protein abundance, identifying post-translational modifications and revealing members of protein complexes at the whole-system level is necessary. Relying on mass spectrometry, coupled with sample fractionation and automated data analysis techniques, we provide such a versatile and powerful data independent acquisition (DIA) platform.
Our Proteomic Quantification Service
Using a high-resolution, fast-sampling mass spectrometer operating in a data-independent mode, we can identify and quantify unknown proteins on a large scale from complex biological samples (such as cultures, tissues, or cells in plasma). Our Discovery Proteomics Platform contains three major data independent quantification strategies that can quantify up to 9,000 proteins per sample under different conditions.
DIA is a high-throughput proteomics research tool that is particularly suitable for clinical biomarker studies. Among the existing DIA technologies, the SWATH (Sequential window acquisition of all theoretical spectra) strategy has been more widely used. DIA can detect all precursor ions, but the selectivity is 5-10 times lower than that of DDA or MRM. The msxDIA technology can increase the selectivity of the precursor ion by 5 times and reduce the complexity of data processing. Compared with the traditional DIA data processing mode, its advantage is that researchers do not need to collect DDA data to build a spectral library. Our Unique Super Reaction Monitoring (SRM™) platform combines Data-Dependent Analysis (DDA) and DIA technologies to obtain data before each analysis. Shortcomings of DDA and selected reaction monitoring (SRM) in terms of depth and width that have limited researchers from examining complex biological systems are addressed by SRM™.
There is no doubt that DIA, which overcomes the randomness of DDA and the low-throughput of MRM, will become a powerful tool for proteome quantification and protein biomarker discovery and verification. However, any technology is not perfect, and it is necessary to choose the applicable quantitative technology according to the actual situation. We will choose the most suitable analysis method for you according to your sample, experimental purpose, etc.
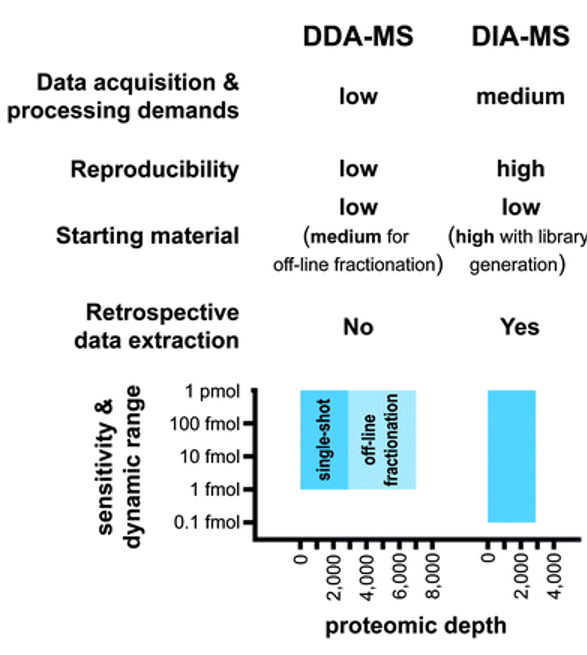 Advantages and limitations of DDA-MS in comparison to DIA-MS[1]
Advantages and limitations of DDA-MS in comparison to DIA-MS[1]
Our Protein Quantification Services Include:
- Enzyme Digestion (Cell/Tissue Protein Extraction and Trypsin Digestion, SDS-PAGE and In-Gel Trypsin Digestion)
- Histone Extraction
- Plasma/Serum Protein Depletion
- Peptide Fractionation
- Peptide Purification and Concentration
- Quality Control Runs
- LC-MS/MS Analysis (DIA Mode)
- Database Search
Advantages:
- High accuracy
- High throughput, more than 9000 proteins can be identified at once
- Quantitatively identify nearly all detectable molecules, covering low-abundance proteins/peptides
- High repeatability
- Complete and comprehensive sample information storage in the first analysis
Sample Requirements:
Based on our special protein extraction technology, we can quickly extract proteins from various samples and design personalized experimental schemes according to different experimental purposes. Specific requirements are as follows:
| Sample Type | Protein | # of Cells | Animal Tissue | Plant Tissue | Blood | Urine | Serum | Microbes |
| Quantify | 100 ug | 1×107 cells | 1 g | 200 mg | 1 mL | 2 mL | 0.2-0.5 mL | Dry weighed: 200 mg |
Note:
- Please prepare enough dry ice or ice packs to ensure low temperature during sample transportation.
- Our service is for research use only and is not intended for diagnosis.
Delivery:
LC-MS/MS Report
Reference:
- Lukas Krasny. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics, 2021, 17, 29-42.
* For Research Use Only. Not for use in the treatment or diagnosis of disease.

 4D Proteomics with Data-Independent Acquisition (DIA)
4D Proteomics with Data-Independent Acquisition (DIA)