Top-down Proteomics in Venom
Venom is usually a complex mixture of chemicals. The advancement of omics technology has deepened our understanding of the venom components related to protein toxins and their role in the chemical-ecological relationship and human health. The rapid development of high-resolution mass spectrometry technology has gradually matured the "top-down" proteomics research. Studying the proteome at the intact protein level can provide more accurate and richer biological information, especially for the case where multiple related post-translational modifications have occurred on the protein.
In addition, due to gene mutations, RNA alternative splicing, and a large number of protein post-translational modifications, the same gene often eventually produces multiple proteoforms. To accurately identify these proteoforms, "top-down" proteomics is indispensable. Separation technology at the protein level, mass spectrometry technology and bioinformatics technology are the three most critical technologies for complete protein identification. Efficient separation technology is the prerequisite for large-scale proteoform identification, effective mass spectrometry fragmentation is the core of providing reliable identification, and fast and accurate mass spectrometry identification algorithms are the guarantee of data analysis efficiency.
Experiment Process
Top-down proteomics uses separation techniques (such as liquid chromatography or 2-D gel electrophoresis) to separate complete proteins from complex biological samples, and then perform MS analysis. Through soft ionization methods such as electrospray ionization (ESI) or matrix-assisted laser desorption ionization (MALDI), ion molecules are generated after fragments are generated through various dissociation methods, such as high energy collision introduction dissociation (HCD), electron capture dissociation (ECD) and electron transfer dissociation (ETD), followed by mass spectrometry.
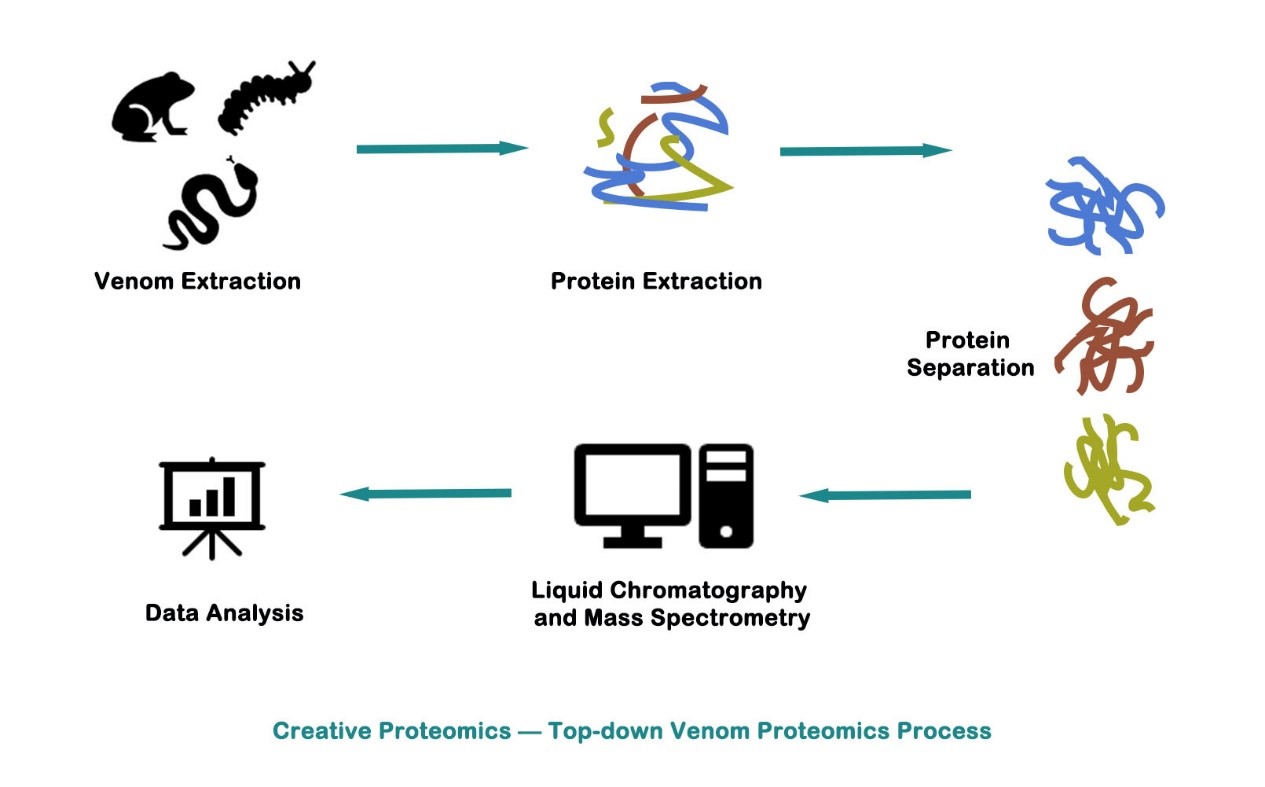
Bottom-up proteomics analysis is a very promising strategy for protein identification, analysis, sequencing and PTM (post-translational modification) characterization. The top-down method allows for the complete protein analysis that has not been enzymatically digested. MS analysis, which means that most of the damaged proteins with unstable structures in the bottom-up analysis strategy can be retained. Therefore, the data of modification sites and modification modes can be obtained at the same time in one spectrum analysis, so as to obtain the relationship data between these modification sites and modification modes. At the same time, compared with the bottom-up method, the protein-free digestion process also reduces the time consumption during the experiment.
Advantages
- Simple sample preparation process
- No need for protein digestion
- Direct detection of protein molecular weight
- A large amount of information is retained, such as PTMs
- Short time consuming
Please contact us for more information.
For research use only. Not intended for any clinical use.
