Novel Proteomic Techniques for Studying Protein Interactions: Proximity Labeling Technology
Protein interactions play a pivotal role in cellular activities, participating in diverse cellular processes across different temporal and spatial dimensions. These include the regulation of the cell cycle, protein synthesis and secretion, signal transduction, and metabolism. Investigating protein interactions is therefore crucial for comprehending molecular regulatory networks. Currently, widely employed methods for studying protein interactions include yeast two-hybrid (Y2H) screening and immunoprecipitation mass spectrometry (IP-MS) analysis. Y2H studies necessitate the construction of suitable cDNA libraries, a time-consuming and labor-intensive step. Additionally, yeast cells, as unicellular eukaryotes, exhibit significant differences from higher eukaryotic multicellular organisms, such as plant cells.
In many instances, immunoprecipitation (Co-IP) has limitations in effectively capturing transient or weak protein interactions. Consequently, scientists have devised a novel proteomic technique for investigating protein interactions known as Proximity Labeling (PL). This innovative approach circumvents the need for library construction or antibody use, offering unique advantages in the identification of hydrophobic and low-abundance interacting proteins, as well as the analysis of weak or transient protein interactions (Yang et al., 2021). First applied in rice protoplasts in 2017, this technique has since found application in related studies in tobacco, Arabidopsis, and tomato. The development and application of Proximity Labeling Technology mark a significant stride in advancing our capabilities to explore intricate protein-protein interactions in diverse cellular contexts.
Select Services
Principles of Proximity Labeling Technology
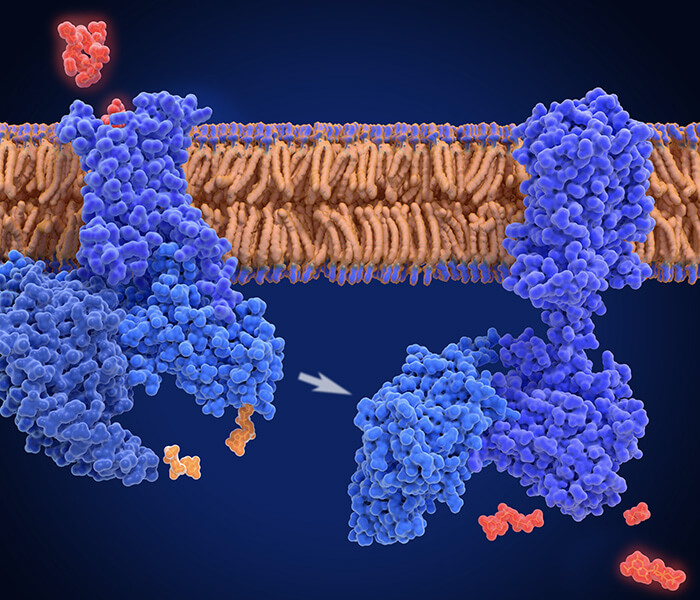
Proximity Labeling is an advantageous technique that can be applied directly to living cells in their native state. This enables the capture of ephemeral or weak protein interactions as they occur in vivo, thereby greatly facilitating the comprehensive investigation of intricate biological processes within cells. Central to this technique is the amalgamation of a probe enzyme with distinct catalytic linking capabilities and the protein of interest.
Through the introduction of diminutive molecule substrates, such as biotin, the link-catalyzing action of the probe enzyme is ignited, which autonomously performs covalent bonding of the diminutive molecule substrate to endogenous proteins situated in close proximity to the said probe enzyme. Post augmented extract enrichment of the labeled proteins, mass spectrometry analysis is executed to distinguish and elucidate the constituent proteins interacting with, or situated in the immediate vicinity of, the target protein. This comprehensive methodology aids researchers in deciphering complex biological processes whilst maintaining native cellular context.
The crux of Proximity Labeling technology lies in the choice of the tool enzyme with catalytic linking activity. Structurally, these enzymes can be broadly categorized into intact and split forms. Intact Proximity Labeling enzymes are primarily utilized for investigating potential interacting proteins of individual target proteins. In contrast, split Proximity Labeling enzymes are employed to explore proteins associated with known protein complexes or interacting proteins (Figure 1). This strategic categorization enhances the versatility and specificity of Proximity Labeling technology, aligning its application with diverse research objectives.
 Fig. 1 Principle of Proximity Labeling technology (Yang et al., 2021).
Fig. 1 Principle of Proximity Labeling technology (Yang et al., 2021).
Commonly Used Proximity Labeling Enzymes
There are numerous enzymes employed for protein-protein interaction identification using proximity labeling techniques. Among them, two frequently utilized classes of tool enzymes are mutants of Escherichia coli biotin ligase (BirA), known as BioID, and Ascorbate Peroxidase (APEX).
The proximity labeling technique based on BioID was the first to be developed and was initially applied in mammalian cells to characterize protein interaction networks (Roux et al., 2012). BioID enzyme biotinylates lysine residues of neighboring proteins. While the enzyme is straightforward and non-toxic, it exhibits relatively low catalytic efficiency, typically requiring 18-24 hours for labeling. Additionally, the optimal catalytic activity temperature for BioID is 37°C, a temperature that induces heat stress in plants. Consequently, this limitation somewhat restricts its widespread application in plant systems.
APEX, developed later, exhibits the ability to biotinylate neighboring proteins within a radius of approximately 20nm of the target protein when supplied with ATP, H2O2, and biotin-phenol. All proteins adjacent to the target, containing tyrosine residues, have the potential to be biotinylated by APEX in just one minute (Rhee et al., 2013). While APEX demonstrates higher catalytic efficiency compared to BioID, its functionality requires the addition of the substrate molecule H2O2. However, the use of H2O2 can induce cellular toxicity, leading to oxidative stress in cells or tissues. Additionally, similar to BioID, APEX operates optimally at a catalytic temperature of 37°C, thereby imposing limitations on its application within plant systems.
In order to enhance the application of proximity labeling technology in plants, the research group led by Alice Ting at Stanford University achieved targeted evolution of BirA through yeast surface display technology in 2018. This effort resulted in the development of a novel proximity labeling enzyme, TurboID (35 kDa), along with its truncated mutant variant, miniTurboID (28 kDa) (Branon et al., 2018). These two innovative enzymes amalgamate the advantages of BioID and APEX, offering high catalytic efficiency without causing damage to live cells.
Notably, TurboID and miniTurboID represent a significant breakthrough in proximity labeling. They demonstrate effective biotinylation at room temperature (25°C), a departure from the requisite 37°C for systems like BioID and APEX. By fusing TurboID with the protein of interest and expressing it within cells, in the presence of ATP and biotin, biotinylation of neighboring proteins within approximately 10nm can be achieved in live cells, with a labeling time of around 10 minutes. Presently, TurboID-based proximity labeling technology has been successfully applied in plant systems, marking a notable stride in advancing the capabilities of proximity labeling in plant biology research.
In conjunction with the previously referenced proximity-labeling enzymes, there has been a continual progressive refinement and evolution of several other enzymes for applications in this field in recent years. This includes enhanced variants such as APEX2 recognized in the APEX series of enzymes, as well as other optimized enzymes that include BioID2, AirID, and BASU, all characterized within the spectrum of the BioID series. Additionally, scientific investigators have unveiled an array of specialized apparatuses including but not limited to HRP, EXCELL, PUP-IT, and NEDDylation which were each specifically tailored for distinct applications. The ongoing advancement and development of these enzymatic tools have substantially lent to and supported the ever-increasing purview and multifaceted nature of proximity-labeling technological methodologies.
Select Services
Table 1 Proximity Labeling technology enzymes currently used for protein interactions identification (Yang et al., 2021).

Primary Applications of Proximity Labeling technology
(1) Protein-Protein Interaction Network Studies
In recent years, proximity labeling technology has increasingly found application in the study of protein-protein interaction networks. This involves fusing proximity labeling enzymes with multiple bait proteins, and integrating mass spectrometry data to obtain corresponding information on proximal proteins. By consolidating the proximity protein groups from various bait proteins and assessing the overlap and intersection, the proximal relationships of core proteins can be determined, leading to the construction of an overarching proximity network (Figure 2).
For instance, Gupta et al. employed 58 bait proteins to delineate the composition and spatial organization of the centrosome-cilium interface, generating a protein interaction network encompassing over 7,000 interactions (Gupta et al., 2015). In a systematic exploration, Anne-Claude Gingras's laboratory investigated the composition of mRNA-related granules and bodies using 139 bait proteins. This effort resulted in a network comprising 1,792 proteins, encompassing 7,424 unique proximal interaction relationships (Youn et al., 2018). Such endeavors showcase the power of proximity labeling technology in unraveling intricate protein-protein interaction networks systematically.
 Figure 2: Employing proximity labeling technology to identify individual protein interactions, decipher spatial relationships among distinct proteins, and construct a protein interaction network. (Yang et al., 2021).
Figure 2: Employing proximity labeling technology to identify individual protein interactions, decipher spatial relationships among distinct proteins, and construct a protein interaction network. (Yang et al., 2021).
(2) Organelle Proteomics and Membrane Studie
Conventional protein studies involve the collection of different organelles, such as the nucleus and mitochondria, through centrifugation-based methods, followed by proteomic analysis. However, due to the lack of techniques to purify specific regions within these organelles, the protein composition of these particular areas cannot be obtained through conventional proteomic methods. Leveraging proximity labeling technology to label proteins in different subcellular regions of organelles allows for further precise separation and elucidation of the protein composition in specific organelle regions.
For instance, the research group led by Alice Ting utilized proximity labeling technology to perform a fine localization analysis of different subcellular regions of mitochondria, including the mitochondrial matrix, outer membrane, and the intermembrane space between the inner and outer membranes (Figure 3A). This approach identified numerous novel mitochondrial proteins and redefined the localization of several mitochondrial proteins that were previously ambiguous or contentious in earlier studies (Rhee et al., 2013; Hung et al., 2014; Hung et al., 2017).
Moreover, proximity labeling technology offers significant advantages in the study of membrane contacts. By fusing split-type proximity labeling enzymes with two organelles that might engage in membrane contacts, the split enzymes reassemble into a complete form upon contact between these organelles. Subsequently, surrounding proteins potentially mediating membrane contacts are labeled, and combining this with mass spectrometry analysis facilitates the identification of proteins playing crucial roles in membrane contacts (Figure 3B). Using this system, scientists have already identified numerous novel proteins associated with endoplasmic reticulum-mitochondria contact sites (Kwak et al., 2020). This exemplifies the substantial potential of proximity labeling technology in advancing our understanding of organelle proteomics and membrane dynamics.
 Figure 3: Utilizing intact or split-type proximity labeling enzymes for proteomic analysis of subcellular compartments (A) and membrane contact sites (B). (Yang et al., 2021).
Figure 3: Utilizing intact or split-type proximity labeling enzymes for proteomic analysis of subcellular compartments (A) and membrane contact sites (B). (Yang et al., 2021).
(3) Protein Topology and Surface Residues
A comprehensive understanding of membrane-localized proteins is essential for unraveling their functional implications in signal transduction, molecular transport, and various cellular processes. The strategic application of proximity labeling technology involves fusing established transmembrane proteins with a proximity labeling enzyme, allowing for the targeted labeling of peptide segments situated on the same side as the proximity labeling enzyme throughout the entire membrane. This, when coupled with mass spectrometry analysis, facilitates the determination of the orientation of membrane proteins, thereby enabling more profound and extensive research (refer to Figure 4A). Notably, scientists have extensively investigated and redefined the membrane topology of numerous proteins localized in the endoplasmic reticulum or mitochondria, leveraging proximity labeling systems based on APEX or Contact-ID (Kwak et al., 2020; Lee et al., 2017; Yoo and Rhee, 2020).
In the realm of protein surface residue analysis, the exposure levels of tyrosine or lysine residues on proteins typically exhibit a positive correlation with the intensity of labeling by the proximity labeling enzyme. Consequently, an in-depth analysis of biotinylation intensity in peptide segments offers valuable insights into the dynamic structural aspects of proteins (refer to Figure 4B). This refined approach enhances our capacity to scrutinize protein surfaces, shedding light on their dynamic interactions within cellular processes.
 Figure 4: Utilizing proximity labeling technology to determine the topological structure and dynamic configuration of proteins. Due to the impermeability of the inner mitochondrial membrane (IMM) to small molecules, proximity labeling occurs exclusively in the intermembrane space (IMS - indicated in red) or on the matrix side (indicated in purple). (Yang et al., 2021).
Figure 4: Utilizing proximity labeling technology to determine the topological structure and dynamic configuration of proteins. Due to the impermeability of the inner mitochondrial membrane (IMM) to small molecules, proximity labeling occurs exclusively in the intermembrane space (IMS - indicated in red) or on the matrix side (indicated in purple). (Yang et al., 2021).
(4) Protein-Nucleic Acid Interactions
The intricate interactions between transcription factors and specific genomic loci play a pivotal role in orchestrating gene expression, contributing significantly to the genomic stability of living cells. Nevertheless, the precise identification of proteins associated with distinct genomic loci in live cells presents a persistent challenge, primarily due to the constraints of conventional methodologies. An effective solution to this challenge emerges through the integration of proximity labeling technology with dCas9.
Despite the loss of DNA cleavage capability, dCas9 maintains its capacity to precisely target specific genomic loci, guided by corresponding sgRNA sequences (Qi et al., 2013). The fusion with dCas9 empowers the proximity labeling enzyme to selectively target predetermined genomic loci, facilitating the subsequent labeling of adjacent proteins (refer to Figure 5A). The proteins labeled in this manner can be systematically enriched and subjected to LC-MS/MS analysis, leading to the identification of proteins intricately associated with specific genomic loci (Gao et al., 2019).
RNA-protein interactions play a vital role in the regulation of gene expression, as well as in mRNA processing, transport, translation, and stability. However, current methods for identifying RNA-associated proteins rely on the purification of RNA-protein complexes and involve in vitro operations, lacking reflection of RNA-protein interactions in the natural cellular environment. The development of proximity labeling technology provides a new solution for studying RNA-associated proteins. For instance, fusing the proximity labeling enzyme with dCas13 allows the investigation of RNA-protein interactions. Guided by sgRNA, dCas13 can specifically target the specific structure of RNA, introducing the proximity labeling enzyme to that site. This allows proteins binding to or near the target RNA sequence to be biotinylated (Figure 5B) (Han et al., 2020).
 Figure 5: By integrating proximity labeling with other existing technologies, identification of proteins binding to specific genomic loci (A) or RNA motifs (B) is achieved. (Yang et al., 2021).
Figure 5: By integrating proximity labeling with other existing technologies, identification of proteins binding to specific genomic loci (A) or RNA motifs (B) is achieved. (Yang et al., 2021).
(5) Subcellular Transcriptome Analysis
In the milieu of eukaryotic cells, RNA often undergoes transportation to designated subcellular compartments. Here, it plays a pivotal role in the orchestration of local translations, a fundamental process that has proven to be an indispensable cog in the machinations of various life processes (Buxbaum et al., 2015). Traditional methodologies employed for investigating subcellular RNA typically encompass techniques such as fluorescence in situ hybridization (FISH), or the amalgamation of immunoprecipitation and either microarrays or high-throughput sequencing. Yet, these methods are not without their downfalls. To elaborate, the inherent biochemical intricacy posed by FISH protocols can often pose quite a formidable challenge, placing them beyond mastery for the average laboratory. Pioneering the advent of proximity labeling technology provides a revolutionary alternative for delving into the exploration of subcellular transcriptome, opening doors towards overcoming past limitations.
For instance, by fusing the proximity labeling enzyme with a specific protein fixed on the endoplasmic reticulum, the surrounding ribosomes can be labeled. Subsequently, biotinylated ribosomes are enriched, and through deep sequencing (i.e., ribosome profiling), the characterized RNA bound to ribosomes can be determined, identifying RNAs undergoing translation (Figure 6). Additionally, proximity labeling technology can be employed to study the non-coding RNA composition in stress granules. Fusion of the proximity labeling enzyme with proteins already reported in stress granules allows for the labeling of surrounding nucleic acids. After isolating and enriching biotinylated nucleic acids, combined with high-throughput sequencing analysis, information about the relevant non-coding RNAs can be obtained (Figure 6).
 Figure 6: Utilizing proximity labeling technology to chart the RNA landscape of membrane-bound or membrane-free organelles. These RNAs encompass both mRNAs engaged in translation and non-coding RNAs. (Yang et al., 2021).
Figure 6: Utilizing proximity labeling technology to chart the RNA landscape of membrane-bound or membrane-free organelles. These RNAs encompass both mRNAs engaged in translation and non-coding RNAs. (Yang et al., 2021).
Case Study
In comparison to its widespread application in animal systems, the utilization of proximity labeling technology in plants, though relatively recent, has yielded numerous research outcomes. Using a selected paper as an example, we will briefly describe the application of proximity labeling technology in studying plant protein interactions.
The pivotal role of NLR (Nucleotide-binding Leucine-rich repeat) immune receptor proteins in mediating plant immunity against a diverse array of pathogens is well-established. Yet, the complexities of the NLR regulatory network and the inherently low expression levels of NLR proteins pose significant challenges to conventional methodologies attempting to capture directly associated proteins in the NLR signaling pathway. On the 19th of July 2019, a milestone research paper titled "TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity" was showcased in the esteemed journal Nature Communications. This study represents a pioneering application of the proximity labeling technology. It centered on investigating the proteins interacting with the N immune receptor protein, a known resistor of the tobacco mosaic virus (TMV). Remarkably, the research identified a previously unknown regulatory factor--UBR7. It was found to interact directly with N NLR and was instrumental in regulating its turnover in the course of TMV resistance. This application of proximity labeling technology offers seminal insights into the underlying apparatus of plant immunity, illuminating a path towards nuanced therapeutic strategies.
The authors established a proximity labeling system in Nicotiana benthamiana (Figure 7) to analyze proteins that may interact with N protein during the immune response against TMV. TurboID was fused to the C-terminus of N protein, with Citrine-fused TurboID serving as a negative control (Figure 8a). After confirming the functionality and unrestricted expression of the fused proteins, the authors analyzed proteins interacting with N protein in the presence or absence of the TMV p50 effector. To quantify the relative abundance of the proximity-labeled proteins, the authors prepared and analyzed Group I samples without p50 and Group II samples with p50 (Figure 8b), ultimately identifying 3198 and 3262 proteins in Groups I and II, respectively.
 Figure 7: Schematic representation of the experimental workflow employing TurboID-based proximity labeling technology in Nicotiana benthamiana. (Zhang et al., 2019)
Figure 7: Schematic representation of the experimental workflow employing TurboID-based proximity labeling technology in Nicotiana benthamiana. (Zhang et al., 2019)
 Figure 8: Identification of proximal and interacting proteins of the N NLR immune receptor (Zhang et al., 2019).
Figure 8: Identification of proximal and interacting proteins of the N NLR immune receptor (Zhang et al., 2019).
Following the in-depth analysis of protein data obtained through mass spectrometry, the authors ultimately identified a specific protein, UBR7, for further investigation. UBR7 functions in the N-terminal arginine-specific proteolysis pathway and is highly conserved across yeast, animals, and plants. Subsequent experiments, including bimolecular fluorescence complementation and GST pull-down assays, provided additional confirmation that UBR7 interacts with N protein. Furthermore, these experiments delineated the interacting region to be the TIR (Toll/interleukin-1 receptor) domain of the N protein (Figure 9).
 Figure 9: Analysis of the interaction between N protein and Nb UBR7 in vivo and in vitro (Zhang et al., 2019).
Figure 9: Analysis of the interaction between N protein and Nb UBR7 in vivo and in vitro (Zhang et al., 2019).
References
- Buxbaum A.R., Haimovich G., Singer R.H. In the right place at the right time: visualizing and understanding mRNA localization. Nat. Rev. Mol. Cell Biol. 2015, 16: 95-109.
- Branon T.C., Bosch J.A., Sanchez A.D., et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36: 880-887.
- Gupta G.D., Coyaud É., Gonçalves J., et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015, 163: 1484-1499.
- Gao X.D., Rodriguez T.C., Sontheimer E.J. Adapting dCas9-APEX2 for subnuclear proteomic profiling. Methods Enzymol. 2019, 616: 365-383.
- Hung V., Zou P., Rhee H.W., et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell. 2014, 55: 332-341.
- Hung V., Lam S.S., Udeshi N.D., et al. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife. 2017, 6: e24463.
- Han S., Zhao B.S., Myers S.A., et al. RNA-protein interaction mapping via MS2- or Cas13-based APEX targeting. Proc Natl Acad Sci U S A. 2020, 117(36): 22068-22079.
- Kwak C., Shin S., Park J.S., et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl. Acad. Sci. U S A. 2020, 117: 12109-12120.
- Lee S.Y., Kang M.G., Shin S., et al. Architecture mapping of the inner mitochondrial membrane proteome by chemical tools in live cells. J. Am. Chem. Soc. 2017, 139: 3651-3662.
- Qi L.S., Larson M.H., Gilbert L.A., et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013, 152: 1173-1183.
- Roux K.J., Kim D.I., Raida M., et al. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196: 801-810.
- Rhee H.W., Zou P., Udeshi N.D., et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013, 339: 1328-1331.
- Youn J.-Y., Dunham W.H., Hong S.J., et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell. 2018, 69: 517-532.e11.
- Yoo C.-M., Rhee H.-W. APEX, a master key to resolve membrane topology in live cells. Biochemistry. 2020, 59: 250-259.
- Yang X., Wen Z., Zhang D., et al. Proximity labeling: an emerging tool for probing in planta molecular interactions. Plant Commun. 2021, 2(2): 100137.
- Zhang Y., Song G., Lal N.K., et al. TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity. Nat Commun. 2019, 10(1): 3252.
You may be interested in: