Significance of Protein Interactions and Interaction Analysis Techniques
Cellular functions are rigorously regulated by protein, nucleic acids, and their interactions, including protein–protein interactions (PPIs), protein-RNA interactions, and protein-DNA interactions. Molecular interaction networks lie at the core of most biological processes, and their dysregulation is associated with various human diseases, including cancer, immune disorders, and neurodegenerative diseases. Thus, elucidating the molecular mechanisms of protein interactions and various complex biological phenomena is of significant importance.
Proteins, as the executors of life activities, often manifest their functions through interactions with other proteins. Studying protein/small molecule-protein interactions plays a crucial role in deepening our understanding and preventing infectious diseases, as well as targeting the treatment of polygenic diseases. Large-scale interactome studies, by analyzing the interactions between individual proteomes, phenotypic characteristics, and environmental factors, reveal the complexity of individual differences and phenotypic diversity and provide insights for therapeutic interventions. This diversity plays an important role in biological evolution, human health, and the development of diseases.
Researchers have developed many methods for studying protein/small molecule-protein interactions, including classical protein interaction research methods and mass spectrometry-based protein interaction research methods. Classical methods mainly include biophysical techniques such as fluorescence resonance energy transfer (FRET), biochemical techniques such as immunoprecipitation, and genetic techniques such as yeast two-hybrid. The low throughput, high noise, and high cost of PPI experiments in classical research methods urgently need to be addressed, leading to the emergence of high-throughput research methods. Mass spectrometry offers advantages such as high sensitivity and high-throughput detection, which can compensate for the shortcomings of classical methods in the study of protein interaction proteomics.
Select Services
Mass Spectrometry-Based Protein Interaction Research Methods
Mass spectrometry (MS)-based proteomics has evolved from characterizing proteins in biological species to assessing protein properties and their functional regulation at multiple levels. With advancements in various aspects of workflow (speed, sensitivity, accuracy, and robustness), it is now possible to perform simplified analyses of almost complete proteomes within a few hours. Fueled by these technological developments, proteome-wide studies of protein interactions have rapidly progressed—enabling the characterization not only of interacting protein networks but also the interrogation of involvement and reactivity of bioactive molecules, elucidation of spatiotemporal dynamics within protein signaling networks, and comprehensive functional annotation of post-translational modifications (PTMs) within the proteome.
Methods for studying protein interactions based on proteomics mainly include affinity purification-mass spectrometry (AP-MS), co-fractionation coupled to mass spectrometry (coFrac-MS), proximity labeling (PL), and cross-linking mass spectrometry (XL-MS), as well as interaction protein screening based on the physicochemical properties of proteins.
Affinity Purification-Mass Spectrometry (AP-MS):
In AP-MS, specific antibodies or other affinity reagents and their potential interacting partners (prey) are selectively purified from cell or tissue lysates. The purified proteins are then identified and quantified by MS, with different baits being used in repeated AP-MS experiments. The statistical calculation of bait-prey pairs from these AP-MS experiments infers protein networks.
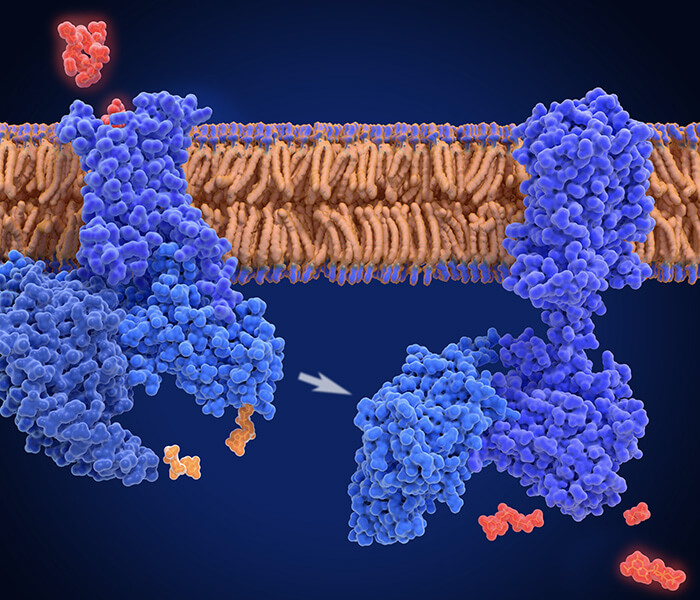
 Strategies for studying interactomes with mass spectrometry (MS) (Zhen et al., 2021).
Strategies for studying interactomes with mass spectrometry (MS) (Zhen et al., 2021).
Co-Fractionation Coupled to Mass Spectrometry (coFrac-MS):
CoFrac-MS is a classical method for studying protein interactions. Spatial-temporal co-behaviors of biomolecules, such as co-expression or co-localization, have been proposed to imply functional or physical interactions. Protein complexes from cell lysates are extensively fractionated under non-denaturing conditions using chromatography or electrophoresis techniques. Each fraction is then digested, analyzed by LC-MS/MS, and analyzed for its protein composition. Subsequently, a hierarchical system of single protein complex subunits can be constructed. Since subunits of intact complexes tend to co-fractionate, biological predictions of protein complexes can be made based on these data, with the correlation between fractions being a core important feature.
Proximity Labeling (PL):
PL is a newly developed and refined method for identifying protein interactions. This method is also known as proximity-dependent biotinylation coupled MS (PDB-MS) or BioID (Proximity- dependent Biotin Identification). This method involves expressing bait proteins fused to biotin ligase enzymes (BioID), horseradish peroxidase (HRP), or ascorbate peroxidase (APEX). The fused enzymes catalyze the conversion of externally added biotin or phenolic biotin into reactive biotin intermediates, which then biotinylate proteins near the bait differently. After biotinylation, cells are lysed, pulled down using streptavidin or neutravidin magnetic beads, and then identified and quantified using MS.
Cross-Linking Mass Spectrometry (XL-MS):
XL-MS is a novel method that combines protein chemical cross-linking techniques with mass spectrometry for studying protein structures and protein interactions. In XL-MS, selected proteins or protein complexes in their native states are first chemically cross-linked with reagents capable of covalently linking spatially proximate amino acid residues. The cross-linked proteins are then digested, and the resulting peptide mixtures are separated and analyzed by LC-MS/MS. Subsequent database searches of the MS data elucidate the sequences of cross-linked peptides and the sites of cross-linking.
Select Services
- Co-immunoprecipitation/mass spectrometry (co-IP/MS)
- Crosslinking Protein Interaction Analysis
- Far-Western Blot Analysis Service
- Pull-Down Assay
- Label Transfer Protein Interaction Analysis
- BioID-MS Service
- Chemical Cross-linking Mass Spectrometry (CX-MS) Service
- Tandem Affinity Purification (TAP)-MS Service
- TurboID Service
- IP-MS Protein Interactomics Solution
Selection Based on Protein Physicochemical Property Changes
Selection based on changes in protein physicochemical properties is a cutting-edge approach that offers a novel and non-conventional method for investigating the dynamics of protein complexes. Unlike traditional techniques that often require structural modifications to the molecules under study, this innovative method enables the exploration of protein interactions by monitoring alterations in key physicochemical properties. These properties include the accessibility to hydrolysis, chemical or thermal stability, and solubility of target proteins.
This approach encompasses a diverse array of methodologies, broadly categorized into four main classes:
Chemically induced changes: This category includes techniques such as Stability of Proteins from Rates of Oxidation (SPROX) and Pulse Proteolysis (PP). SPROX involves measuring the stability of proteins by assessing their susceptibility to oxidation, providing valuable insights into their structural integrity and interactions. PP, on the other hand, utilizes proteolytic enzymes to probe protein conformational changes and complex formation dynamics.
Heat-induced changes: Methods falling under this classification include Cellular Thermal Shift Assay (CETSA) and Thermal Proteome Profiling (TPP). CETSA exploits changes in protein thermal stability upon ligand binding or complex formation, offering a robust approach for studying protein interactions in cellular environments. TPP, on the other hand, involves subjecting cells or lysates to a series of temperature shifts, followed by mass spectrometry analysis to map temperature-dependent changes in protein abundance and stability.
Proteolysis resistance-based methods: This category encompasses techniques such as Drug Affinity Responsive Target Stability (DARTS) and Limited Proteolysis-Mass Spectrometry (LiP-MS). DARTS relies on the differential protease susceptibility of protein targets in the presence or absence of interacting molecules, providing insights into ligand-induced conformational changes and protein-protein interactions. LiP-MS combines limited proteolysis with mass spectrometry analysis to identify protease-resistant protein fragments, enabling the mapping of protein interaction interfaces and conformational changes.
Solubility-dependent methods: SIP (Solubility-Dependent Methods) leverage changes in protein solubility upon interaction with binding partners or ligands. By monitoring alterations in protein solubility under different experimental conditions, these methods offer valuable insights into protein complex formation, stability, and dynamics.
Reference
- Zhen, C. H. E. N., and C. H. E. N. Junjie. "Mass spectrometry-based protein‒protein interaction techniques and their applications in studies of DNA damage repair." Journal of Zhejiang University. Science. B 22.1 (2021): 1.
You may be interested in: