Structure and pathogenic mechanism of SARS-CoV-2
The COVID-19 pandemic was triggered by a new type of highly pathogenic coronavirus, known as SARS-CoV-2.
The genome of SARS-CoV-2 is similar to other coronaviruses and consists of four key structural proteins: spike protein (S), envelope protein (E), membrane protein (M), and nucleocapsid protein (N).
The S protein is a class I fusion protein with an extracellular domain, a transmembrane domain, and an intracellular tail. The highly glycosylated extracellular domain protrudes from the surface of the virus envelope and promotes attachment and fusion with the plasma membrane of the host cell. The extracellular domain can be subdivided into host receptor-binding domain (S1) and membrane fusion (S2) subunits, produced by proteolysis of host protease at S1/S2 and S2' positions. After being cut, the S1 and S2 subunits remain associated and assemble into a coronal homotrimer.
In humans, S proteins from both SARS-CoV and SARS-CoV-2 use angiotensin-converting enzyme 2 (ACE2) protein as a receptor for cell entry. The subsequent host response to SARS-CoV-2 infection involves a complex signal cascade, which can be observed by perturbing protein phosphorylation patterns, leading to the production of interferons and cytokines. During this signal transmission process, SARS-CoV-2 triggers the response of the TLR-IRAK pathway in host cells, and further mediates the production of interferons and cytokines. Cytokines such as IL6 and IL8 often transmit signals through JAK-STAT and other pathways in host cells to amplify the host immune response.
Application of proteomics in SARS-CoV-2 research
The research article titled "The Global Phosphorylation Landscape of SARS-CoV-2 Infection" was published in the journal Cell last year. It is collaborative research among University of California, San Francisco, and other institutions. This article analyzed the systemic phosphorylation modifications caused by the new coronavirus infecting cells and discovered new potential drug candidates for abnormal phosphorylation pathways.

This study uses SARS-CoV-2 infection in Vero E6 cells as a model. Six time points of infection were designated at 0h, 2h, 4h, 8h, 12h, and 24h. As a result, 4,624 phosphorylation sites and 3,036 proteins were identified. Results demonstrated that the level of phosphorylation changed more significantly between different time points, reflecting the importance of phosphorylation in the rapid infection process.
The results of the phosphorylation group showed that some viral proteins could be phosphorylated. Through sequence analysis and prediction, the host's CK2, CDK, and PKC were found to be the upstream kinases that lead to the phosphorylation of these viral proteins. Researchers speculate that these kinases may play the role of "accomplices" in the virus infection. At the host level, the prediction of the phosphorylation site cluster analysis showed that the kinase activities of p38MAPK, CK2, and CaMK2G were up-regulated, while the kinase activities of CDK, Akt, and Rho families were down-regulated.
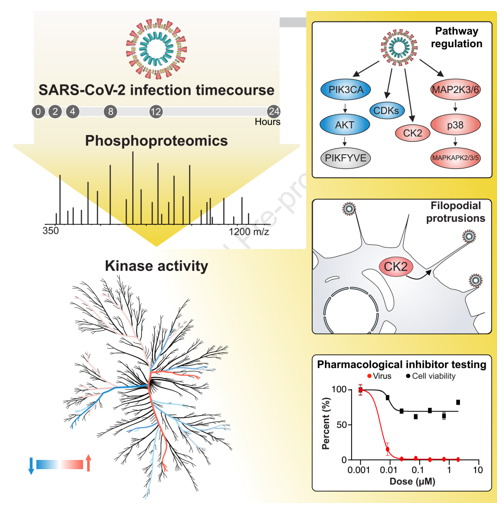
Studying these differentially expressed kinases may find potential drug treatment targets, providing new ideas and basis for future treatment.
Creative Proteomics can provide you with proteomics, revealing information about protein abundance, variation and modification and their interacting partners and networks. Our services include:
Protein identification service
Protein quantification service
Protein post-translational modification analysis service