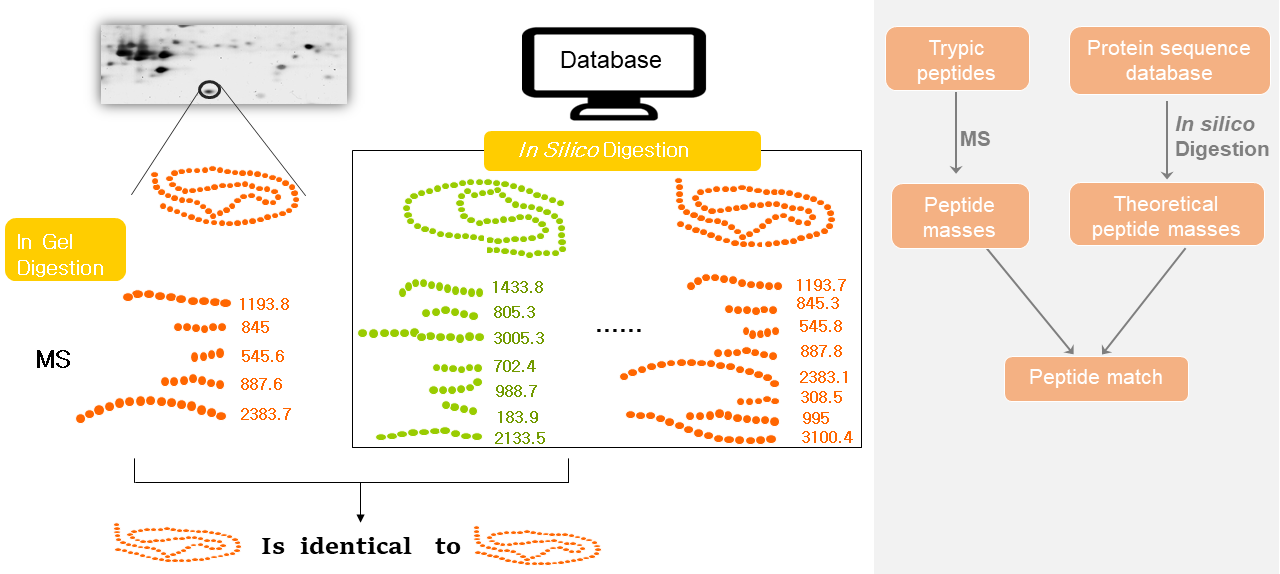
Peptide Mass Fingerprinting (PMF), also known as mass fingerprinting, was developed in 1993. It is a high throughput protein identification technique in which the mass of an unknown protein can be determined. PMF is always performed with Matrix-assisted laser/desorption ionization time of flight (MALDI-TOF) mass spectrometry.
In this phrase "peptide mass fingerprinting", peptide means protein fragment, which is often generated by trypsin. Mass means the molecular size of peptides. And the fingerprinting presents the uniqueness of the masses of peptides. It means that the digestion of a protein by an enzyme can provide a specific fingerprint of great specificity, which can possibly identify the protein from this information alone.

In this technique, after separation of proteins by gel electrophoresis or liquid chromatography and being cleaved with a proteolytic enzyme, we can get experimental peptide mass through mass spectrometry. On the other hand, the theoretical masses are achieved by using computer programs that translate the known genome of the organism into proteins or the proteins in the database, then theoretically cut the proteins into peptides, and calculate the absolute masses of the peptides. The experimentally obtained peptide masses are compared with theoretical peptide mass. The results are statistically analyzed to find the best match.
There are several steps for PMF:
1. Protein separation. The proteins of interest from a sample are separated by gel electrophoresis, usually with 2D PAGE.
2. Digestion. The protein of interest is digested by the proteolytic enzyme. Trypsin is the favored enzyme for PMF. It is relatively cheap, highly effective and generates peptides with an average size of about 8-10 amino acids, which is suitable for MS analysis. The endoprotease Lys-C, Glu-C (V8) or other endoproteases can be used for specific needs.
3. Mass Spectrometric analysis. The peptides can be analyzed with different types of mass spectrometers, such as MALDI-TOF or ESI-TOF. MALDI-MS, which is fast, sensitive, accurate and can be automated, is the most commonly used technique to perform PMF for the reason that it allows higher sample throughput and several proteins can be analyzed in a single experiment. The mass spectrometric analysis produces a peak list, which is a list of molecular weights of the fragments.
4. In silico digestion. Software performs in silico digestion on database proteins with the same enzyme used in the experimental digestion and generates a theoretical peak list. Mascot, MS-Fit, and Profound are the most frequently used search programs for PMF.
5. Comparison. Compare peak list and theoretical peak list to get best match.
PMF is easy to perform and no need for too much optimization. It is significantly faster to carry out than peptide sequencing and in this method, only the masses of the peptides is needed to be known. Although PMF has some advantages, it dose have some disadvantages. The protein sequences of interest need to be present in the database. And PMF fails to identify the mixture protein which can complicate the analysis and compromise the results. What’s more, peptides containing post-translational modifications may not be correctly identified.
PMF by MALDI-MS play an important role in the identification of well-separated proteins. The development of mass spectrometry and the rapid growth of databases have revolutionized the identification of proteins.
Reference:
Thiede B, Höhenwarter W, Krah A, et al. Peptide mass fingerprinting. Methods, 2005, 35(3): 237-247.