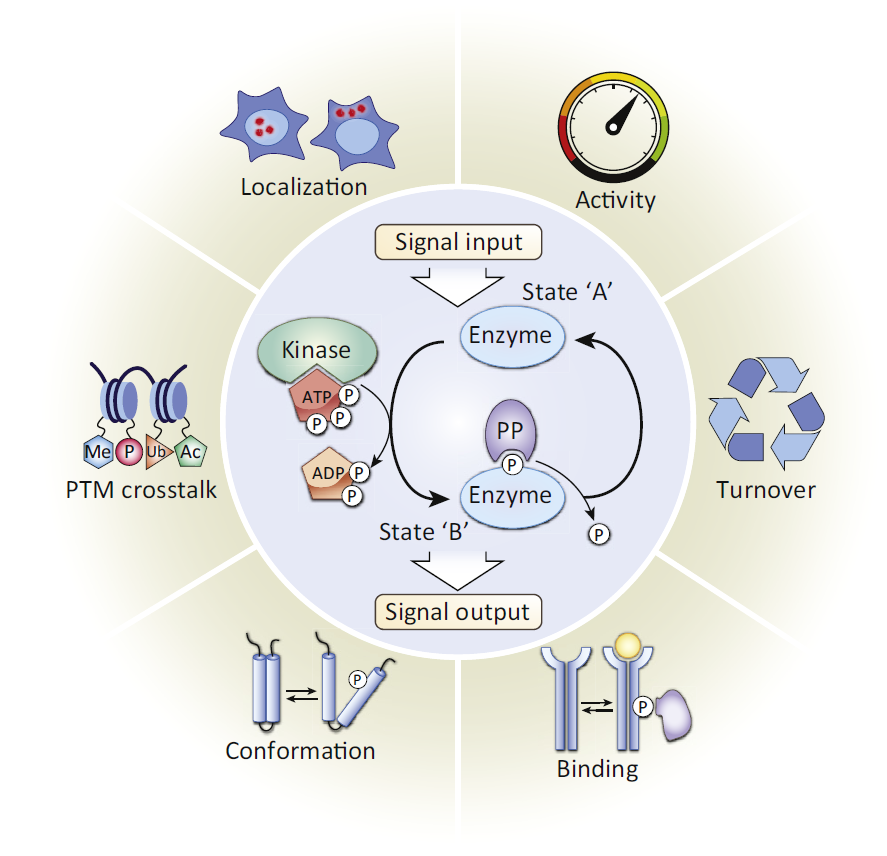
Protein phosphorylation, a reversible process, is characterized by adding phosphate donated from ATP and removing phosphate from a phosphorylated protein substrate. And the protein phosphorylation is catalyzed by protein kinase and phosphatase (PP). In eukaryotic cells, about 86.4% of the protein phosphorylation events occur on serine (Ser or S), while 11.8% protein phosphorylation events occur on threonine(Thr or T) whereas only 1.8% on tyrosine (Tyr or Y) residues.
Protein phosphorylation plays important roles in many cellular processes, including cell cycle, growth, apoptosis, and signal transduction pathways. In addition, phosphorylation can modulate enzyme activity, alter its affinity to other proteins, and transmit signals through kinase cascades that often are branched and interactive. In order to understand these mechanisms, it is necessary to analyze protein phosphorylation. There are many methods for phosphorylation analysis, including kinase activity assays, phosphor-specific antibody development, western blot, enzyme-linked immunosorbent assay (ELISA), as well as Intracellular Flow Cytometry and immunocytochemistry/immunohistochemistry (ICC/IHC). In addition to these methods, MS analysis plays an important role in phosphorylation analysis. There are two common strategies for identifying protein phosphorylation sites through bottom-up proteomics, including identifying phosphorylation sites from individual proteins and small protein complexes, and identifying global phosphorylation sites from the whole cell and tissue extracts.
Single protein (protein complex) phosphorylation site mapping
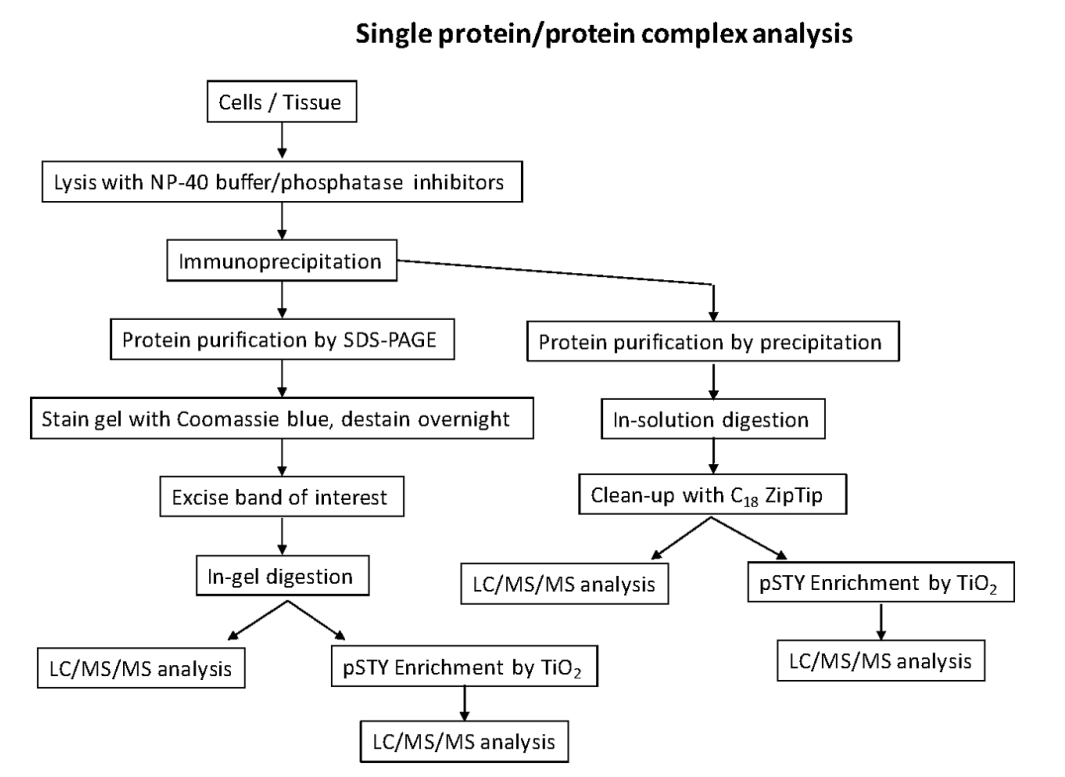
Identifying phosphorylation sites of a specific protein of interest is important for comprehensively study the functional role of phosphorylation from the protein. There are several steps.
- Sample preparation. Because of the low stoichiometric level of phosphorylated peptides compared to unmodified peptides, there is a need to purify as much protein as possible for successful liquid chromatography-tandem mass spectrometry (LC-MS/MS). This is typically done through an epitope tag such as FLAG, HA or Myc or through immunoprecipitation (IP) with a suitable antibody. In addition, protein purification by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) or by precipitation is also available. To enrich phosphorylated peptides, there are several methods available, including metal ion affinity chromatography (IMAC) and titanium dioxide beads (TiO2).
- Analysis by LC-MS/MS. The peptides can be introduced into the mass spectrometer through liquid chromatography coupled with electrospray ionization (ESI), or matrix-assisted laser/desorption ionization (MALDI). Peptides are most commonly sequenced by MS/MS using collision-induced dissociation (CID), collisions with an inert gas cause the protonated peptides to fragment at the amide bonds along the backbone. Electron capture dissociation (ECD) and Electron transfer dissociation (ETD) can be applied for phosphorylation analysis.
- Data analysis. Informatics software is necessary to identify peptide sequences and their phosphorylated counterparts. Some database search engines are available, including Mascot, Sequest, MaxQuant, and X!Tandem.
Global analysis of protein phosphorylation by mass spectrometry
The advent of methods for enrichment of phosphoproteins in conjunction with improvements in mass spectrometry has resulted in the identification of large numbers of phosphoproteins and phosphosites. With the advancement of MS instrumentation that allows for the rapid and sensitive sequencing of peptides and the concurrent development of phosphopeptide enrichment strategies, we now have the ability to analyze changes in protein phosphorylation at an unprecedented scale. From hundreds of studies, more than 300,000 phosphorylation sites have been identified across a multitude of both prokaryotic and eukaryotic species, with more than 200,000 of these coming from mammals alone. Greater than 95% of these sites have been identified using MS-based phosphoproteomics.
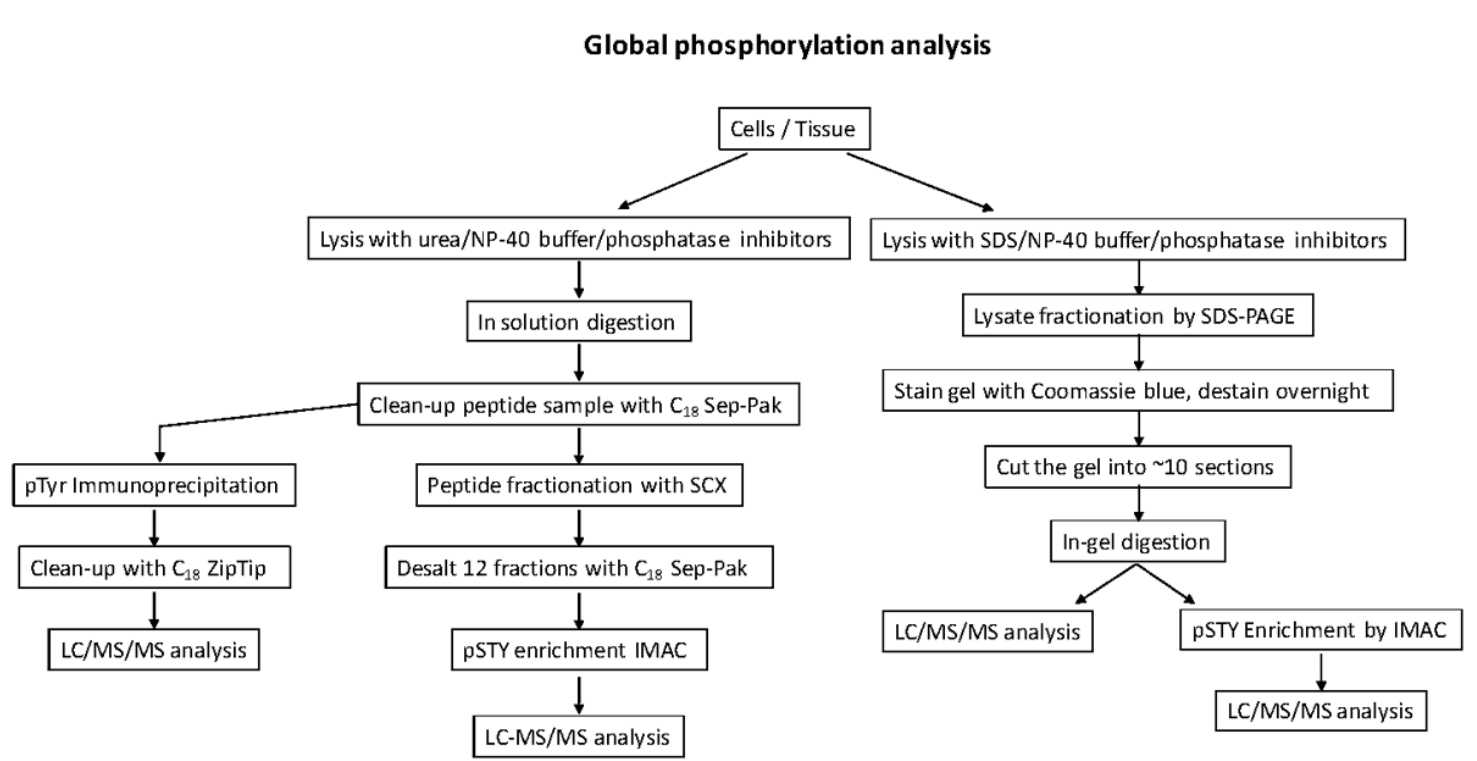
Global phosphorylation site analyses that capture pSer/pThr/pTyr sites from biological sources sites are more resource and time-consuming. It has several strategies.
- This strategy involves digesting the whole cell lysate, followed by peptide fractionation by strong cation exchange chromatography (SCX), phosphopeptide enrichment by IMAC or TiO2.
- In this strategy, the protein lysate can be fractionated by SDS-PAGE, followed by digestion, phosphopeptide enrichment and LC-MS/MS.
- In addition, one can also IP only phospho-tyrosine peptides using a pTyr antibody followed by LC-MS/MS.
For a truly comparative analysis of global phosphorylation, a quantitative proteomics strategy is necessary. Quantitative phosphoproteomics allows researchers to investigate signaling pathways in different model systems to identify phosphorylation events that vary in terms of abundance and duration as a result of a given stimulation.
Equipped with advanced facilities, Creative Proteomics can provide phosphorylation services in an affordable manner. Our team of experts with extensive experience can help you understand what you are trying to investigate and give you the most appropriate solutions.
References:
- Humphrey, Sean J., David E. James, and Matthias Mann. "Protein phosphorylation: a major switch mechanism for metabolic regulation." Trends in Endocrinology & Metabolism26.12 (2015): 676-687.
- Breitkopf, Susanne B., and John M. Asara. "Determining in vivo phosphorylation sites using mass spectrometry." Current protocols in molecular biology(2012): 18-19.
- McNulty, Dean E., Timothy W. Sikorski, and Roland S. Annan. "Identification and analysis of protein phosphorylation by mass spectrometry." Analysis of Protein Post-Translational Modifications by Mass Spectrometry(2016): 17-87.