Protein-protein interactions (PPIs) are the basis of the cellular structure and functions. It has been estimated that more than 80% of proteins do not operate alone but in complexes. The analysis of PPIs is able to help us understand cellular organization, process, and functions. PPIs analysis may discovery unique and unforeseen functional roles for well-known proteins. And it can discover previously unknown proteins by association knowns proteins.
There are large numbers of approaches to investigate PPIs, and each of the methods has its own strengths and weaknesses. These PPI detection methods can be classified into three groups, including in vitro, in vivo, and in silico methods. To validate, characterize and confirm PPIs, we often combine different techniques. Since the methods to investigate PPIs are too numerous to describe here, we focus on some common methods.
Common methods to analyze protein-protein interactions
Co-immunoprecipitation (Co-IP)
Immunoprecipitation of intact protein complexes is recognized as co-immunoprecipitation. In Co-IP, the proteins are present in their native form in a complex mixture of cellular components. As for its principle, targeting a known protein with a specific antibody can pull the entire protein complex, therefore, the unknown members of the complex can be identified. It is suitable for proteins involved in the complex bind to each other tightly. Immunoprecipitation experiments reveal direct and indirect interactions, so results may indicate that two proteins interact directly or may interact via one or more bridging molecules.
Pull-down assay
The pull-down assay is a common variation of co-IP, which can be used to determine a physical interaction between two or more proteins and to identify previously unknown PPIs as an initial screening assay. In a pull-down assay, a purified and tagged protein as a bait protein to bind any interacting proteins is immobilized on an affinity ligand specific to the ligand. The common tags include GST-tag, His-tag, and biotin-tag. When the cell lysate is incubated with an immobilized bait protein, proteins binding to the bait protein can be captured and “pulled down”.
Label transfer assay
Laber transfer has been widely used for screening or confirming PPI. In fact, label transfer is very valuable to identify transient or weak PPIs that are difficult to capture using other in vitro detection strategies. In this method, the protein of interest is tagged with a label transfer reagent (LTR, composed of a label radical and a photoreactive group). After the crosslinking reaction, the label is transferred to an interacting partner and allowing its determination by multiple methods, like western blot analysis, protein sequence analysis, and mass spectrometry.
Methods for proteome-scale interactome maps
Mapping protein-protein interactions on a proteomics scale reveal the macromolecular connections that underlie the biology of the cell. Although there are many methods for detection PPIs, only a few methods are able for high-throughput mapping. Binary mapping by yeast two-hybrid (Y2H) and co-complex associations affinity purification followed by mass spectrometry (AP-MS) or co-fractionation with mass spectrometry (CO-FRAC) can be adapted to systematically survey the entire proteome, each technique has inherent benefits and limitations.
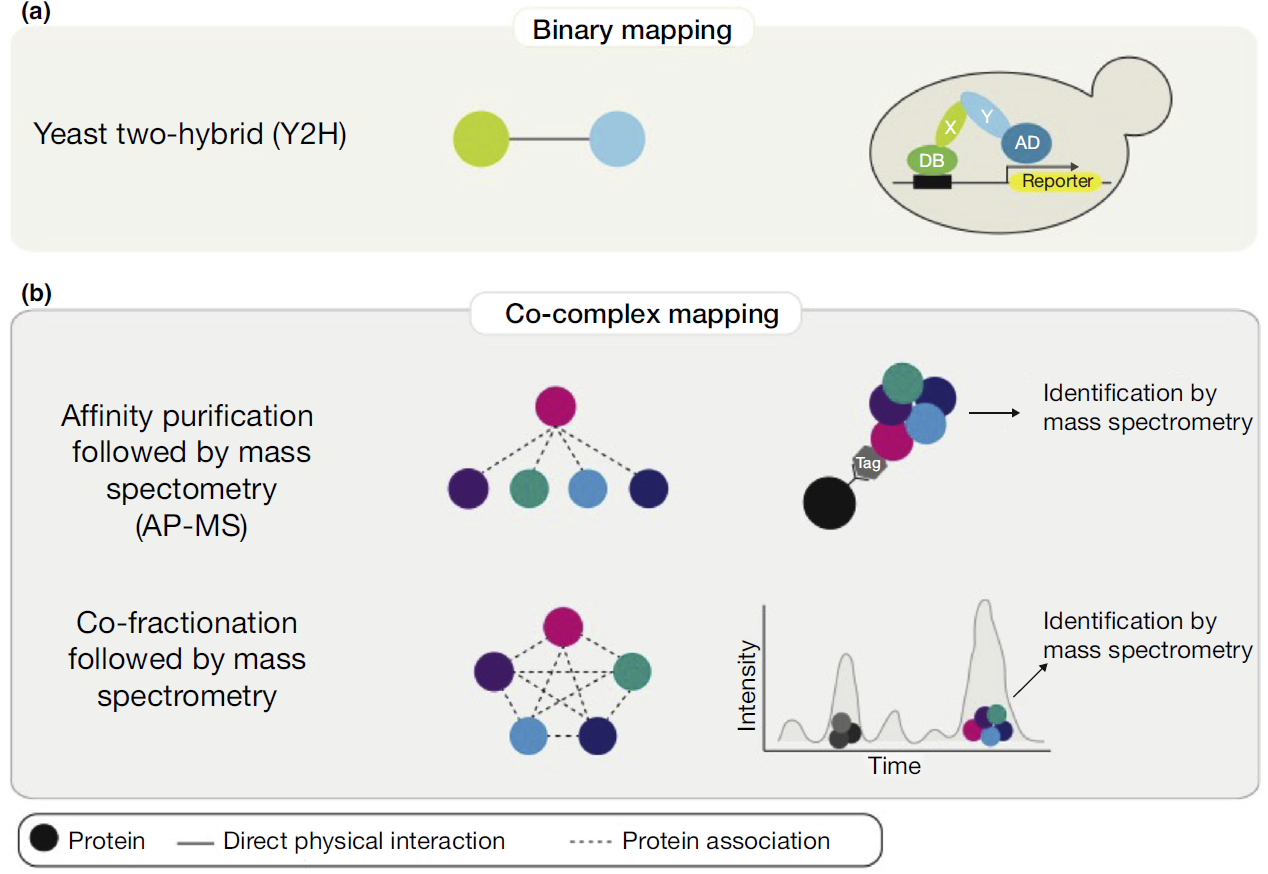
Figure 1. Schematic of systematic experimental methods for high-throughput proteome-scale mapping of protein-protein interactions. ( Cafarelli T M et al., 2017)
- Binary mapping by yeast two-hybrid. It detects direct physical interactions between two proteins by the reconstitution of a transcription factor that activates reporter gene expression and promotes yeast cell survival on appropriate selective media.
- Affinity purification and mass spectrometry. In this method, epitope tags are fused to bait proteins, and proteins associated with the bait proteins are purified and identified by mass spectrometry.
- Co-fractionation and mass spectrometry. It doesn’t need exogenously introduced ORFs or protein tags. Chromatographic and other biochemical separation methods of cell extracts are carried out to generate hundreds to thousands of fractions that are analyzed by mass spectrometry.
Protein-Protein Interaction Databases
Protein-protein interaction can generate the massive quantity of experimental data. Therefore, it is a need to construct computer-readable biological databases in order to organize and to process data. In the following table, we introduce some commonly used protein-protein interaction databases.
Table 1. Commonly used protein-protein interaction databases
| Name | URL | Description |
| BioGRID | http://thebiogrid.org/ | BioGRID is an interaction repository that archives and disseminates genetic and protein interaction data from model organisms and humans, which currently holds over 1,400,000 interactions curated from both high-throughput datasets and individual focused studies |
| DIP | http://dip.doe-mbi.ucla.edu/dip/Main.cgi | The DIP (database of interacting proteins) lists protein pairs that are known to interact with each other, which combines information from a variety of sources to create a single, consistent set of protein-protein interactions. |
| HitPredict | http://hintdb.hgc.jp/htp/ | HitPredict is a resource of experimentally determined protein-protein interactions, which currently includes 12 species (like Arabidopsis thaliana, Escherichia coli, Homo sapiens, Saccharomyces cerevisiae, etc) |
| IntAct | https://www.ebi.ac.uk/intact/ | IntAct, a freely available and open source database system and analysis tools for molecular interaction data, provides PPI information derived from literature curation or direct user submissions, |
| Interactome3D | http://interactome3d.irbbarcelona.org/ | Interactome3D can provide the structural annotation of PPI networks. |
| Mentha | http://mentha.uniroma2.it/ | Mentha archives evidence collected from different sources, which offers eight interactomes. |
| Pathway commons | https://www.pathwaycommons.org/ | Pathway Commons is a web resource for collecting and disseminating biological pathway and interaction data. |
| STRING | http://string-db.org/ |
STRING is a database of known and predicted protein-protein interactions, including direct (physical) and indirect (functional) associations. |
At Creative Proteomics, our team of experts with extensive experience can help you understand what you are trying to investigate and give you the most appropriate solutions about protein and protein interaction. We can provide protein-protein interaction services based on different methods, including:
- Co-immunoprecipitation (co-IP)
- Pull-Down Assay
- Crosslinking Protein Interaction Analysis
- Label Transfer Protein Interaction Analysis
- Far-Western Blot Analysis
References:
- Cafarelli T M, et al. Mapping, modeling, and characterization of protein-protein interactions on a proteomic scale. Current opinion in structural biology, 2017, 44: 201-210.
- Rao V S, et al. Protein-protein interaction detection: methods and analysis. International journal of proteomics, 2014, 2014.