Peptidomics can be easily defined as the study of the low molecular weight fraction protein fragments from endogenous protein degradation products, or other small proteins such as cytokines and signaling peptides. Peptides in the peptidome can be classified into two parts. One is the bioactive peptides shed from all cell types in the microenvironment, which can serve as reporters for cell-to-cell communications, such as hormones and cytokines. The other is peptide fragments cleaved by enzymes resulting from in vivo resident proteins.
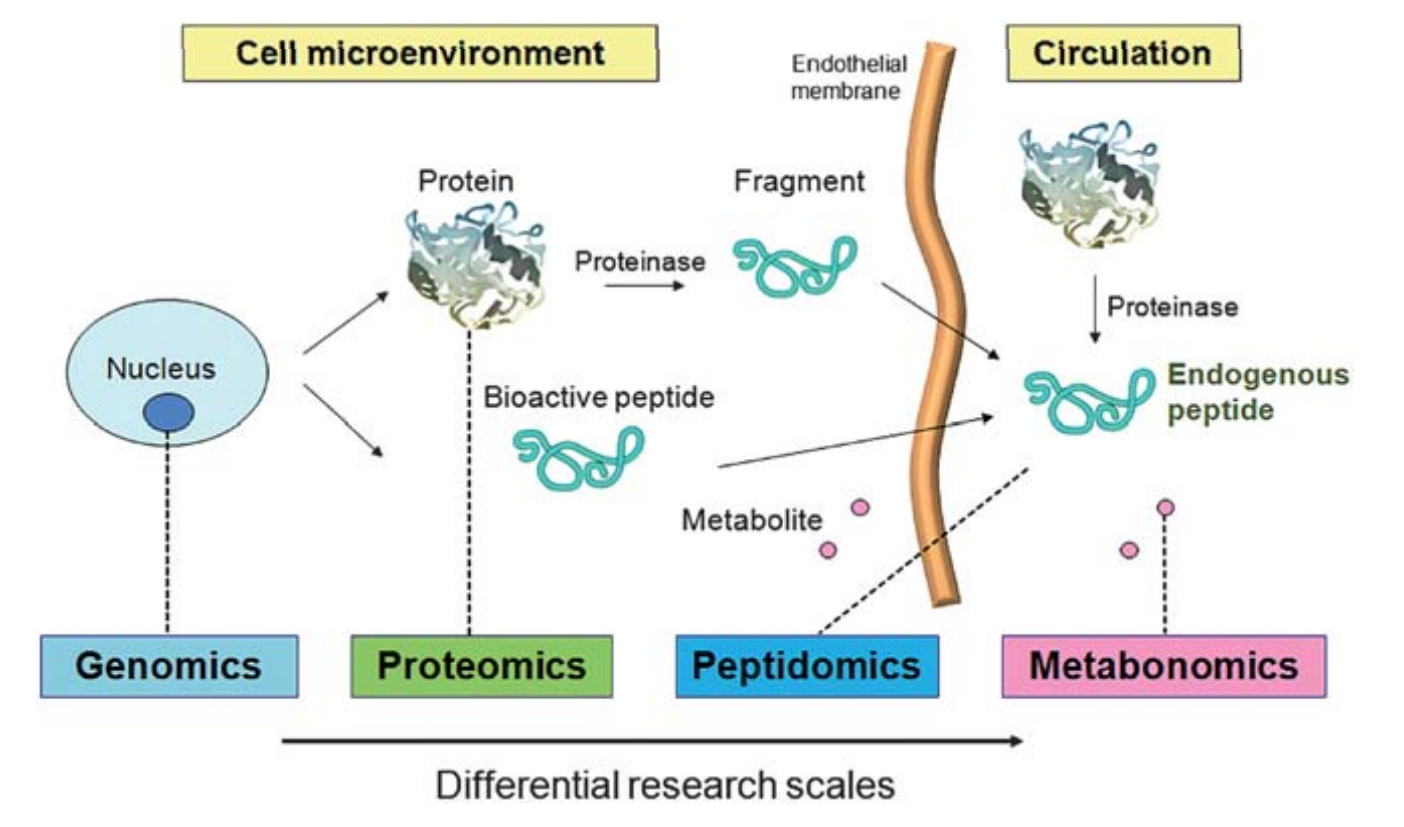
Figure 1. The term peptidome refers to two types of in vivo peptides. (Gao Y, et al, 2011)
Biomarkers are molecules that indicate a physiological state and also the change during a disease process. The usability of a biomarker lies in its ability to provide an early indication of the disease, to monitor disease progression, to provide ease of detection, and to provide a factor measurable across populations. Due to their multiple involvements in biological systems, peptides are ideal biomarkers.
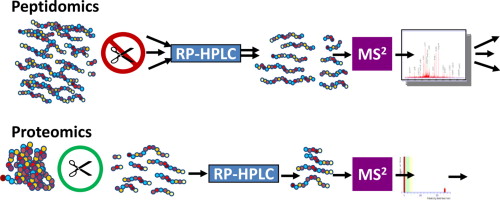
In human bodies, peptidome biomarkers can be used to forecast disease, diagnose various disorders, guide clinical therapy, and monitor medicine response. The mass spectrometry-based peptidomics for biomarker discovery contains sample preparation, separation, detection and identification, quantitative evaluation, data analysis, as well as biomarkers validation.
Sample Preparation
The biomarker discovery research in biologic samples is usually hampered by the vast complexity and large dynamic range of the proteins. Because many disease identifying biomarkers are likely to be low-abundance proteins, it is imperative to remove the high-abundance proteins or apply enrichment techniques to allow detection and better coverage of the low-abundance proteins for MS analysis. Many methods and technologies have been applied to extract peptides and small proteins, such as organic solvent precipitation, ultrafiltration, magnetic beads, solid-phase extraction (SPE), affinity removal column, strong cation exchange (SCX), etc. In addition, ultrafiltration is realized by filtering the sample using a membrane of a defined molecular weight cut-off (MWCO) based on size exclusion mechanism
Separation
Before being injected into the mass spectrometer, the peptides need to be separated because of the high complexity. For example, when using MS, the peptides with the same amino acid composition but different sequences generate the same mass spectra of molecular ions. As a technique usually combined with MS, liquid chromatography (LC) is the most popular method for peptide separation. And high-performance liquid chromatography (HPLC) has been widely used for bioactive peptide profiling. In addition, there are several other methods, including multidimensional separation technology, and capillary electrophoresis (CE).
Detection and Identification
Detection of numerous fragments necessitates the development of high-throughput, accurate, and sensitive mass spectrometer. There are four commonly used types of mass analyzers: quadrupole, time-of-flight, ion trap, and Fourier transform ion cyclotron resonance. These analysers are usually put together in tandem to strengthen their superiorities. In order to determine the amino acid sequence of a specific peptide, the tandem MS mode should be chosen in the operation. The collision-induced dissociation (CID) fragmentation technique is incorporated in basically all types of tandem MS. Electron capture dissociation (ECD) and electron transfer dissociation (ETD) are developed for providing explicit fragmental spectra.
Quantitative Evaluation
A detection system for peptide biomarkers has two tasks. One is to show which peptides are present in a sample (qualitative analysis). The other is to determine the concentration (quantitative analysis). With the development of the mass spectrometry, there are various techniques for gel-free quantitative analyses of peptidomics samples. The non-gel quantification can be divided into two main categories: stable isotope labeling and label-free methods. Various isotope labeling methods include ICAT, TMT, iTRAQ and so on. But it has some limitations, like the increasing cost, complexity of sample preparation, complicate data processing, etc. Currently, label-free quantitative approaches have been developed based on the chromatographic peak area of a peptide or frequency of MS/MS spectra correlating to the peptide concentration.
Data Analysis
The mass spectrometer can produce a large amount of data, which is complex and hard to analyze. The data is submitted to data mining algorithms to get the statistical interpretation, which is divided into two categories, unsupervised methods and supervised methods. Mass spectra are analyzed using search engines. There are some commonly used search engines, including Mascot, SeQuest algorithms, X!Tandem. Peptide sequencing information databases include non-redundant NCBI, Erop-Moscow, Peptidome, Pepbank and so on.
Biomarkers Validation
After biomarker discovery and characteristics, there is a need to validate putative biomarkers identified by the MS-based analysis. It requires to offer orthogonal analysis to rule out a false positive by MS and providing additional evidence for the biomarker candidates from the study for future potential clinical assays. Nowadays, antibody-based assays (like Western blotting, ELISA, and immunochemistry) are commonly used methods for biomarker validation. In addition, quantitative assays based on multiple-reaction monitoring (MRM) MS as an attractive alternative, have been employed in biomarker verification due to its enhanced throughput and specificity.
References:
- Cunningham R, et al. Mass spectrometry-based proteomics and peptidomics for systems biology and biomarker discovery. Frontiers in biology, 2012, 7(4): 313-335.
- Gao Y, et al. Peptidome workflow of serum and urine samples for biomarker discovery. Analytical Methods, 2011, 3(4): 773-779.