De novo is Latin which means "over again" or "anew". The de novo peptide sequencing is a method for peptide sequencing performed without prior knowledge of the amino acid sequence. This method can obtain the peptide sequences without a protein database, which can overcome the limitations of database-dependent methods like peptide mass fingerprinting (PMF). It uses computational approaches to deduce the sequence of peptide directly from the experimental MS/MS spectra. It can be used for un-sequenced organisms, antibodies, peptides with posttranslational modifications (PTMs), and endogenous peptides.
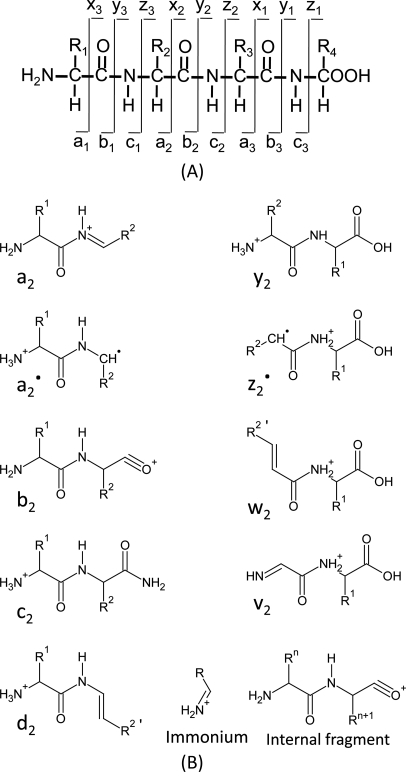
In this method, the peptide is fragmented along the peptide backbone and the resulting fragment ions are measured to produce spectra. There are 3 ways to break bonds to form peptide fragment: alkyl carbonyl (CHR-CO), peptide amide bond (CO-NH), and amino alkyl bond (NH-CHR). Therefore, it can form 6 types of fragmentation ions, including the N-terminal charged fragment ions which are classed as a, b, or c, and the C-terminal charged ones which are classed as x, y, or z. And because the peptide amide bone (CO-NH) is the most vulnerable, the most common peptide fragments observed in low energy collisions are a, b and y ions. De novo methods use the knowledge of the fragmentation methods employed in the MS. CID, Collision induced dissociation, also known as collisionally activated dissociation, is the most common form of fragmentation. In this method, The ions can obtain high kinetic energy and collide with neutral molecules. Some of the kinetic energy is converted into internal energy which leads to bond breakage and the fragmentation of the molecules into smaller fragments. This method results in the formation of b and y series ions from the precursor ion. The Electron capture dissociation (ECD) and Electron transfer dissociation (ETD) have been implemented in the recent mass spectrometer. In these methods, Ions are fragmented after reaction with electrons. After fragmentation, it forms c and z type ions through cleavage of the peptide bond between the amino group and alpha carbon.
The mass can usually uniquely determine the residue. The main principle of de novo sequencing is to use the mass difference between two fragment ions to calculate the mass of an amino acid residue on the peptide backbone. For example, the mass difference between the y7 and y6 ions in the following figure is equal to 101, which is the mass of residue T. Thus, if one can identify either the y-ion or b-ion series in the spectrum, the peptide sequence can be determined. However, the spectrum obtained from the mass spectrometry instrument does not tell the ion types of the peaks, which need an expert or a computer algorithm to figure out. There are couples of software packages used for de novo sequencing, such as PEAKS, Lutefisk, PepNovo, SHERENGA, etc. But there are some notes you have to mind. y and b ion fragments which contain the amino acid residues R,K,Q, and N may appear to lose ammonia (-17). Y and b ion fragments which contain the amino acid residues S, T, and E may appear to lose water (-18).
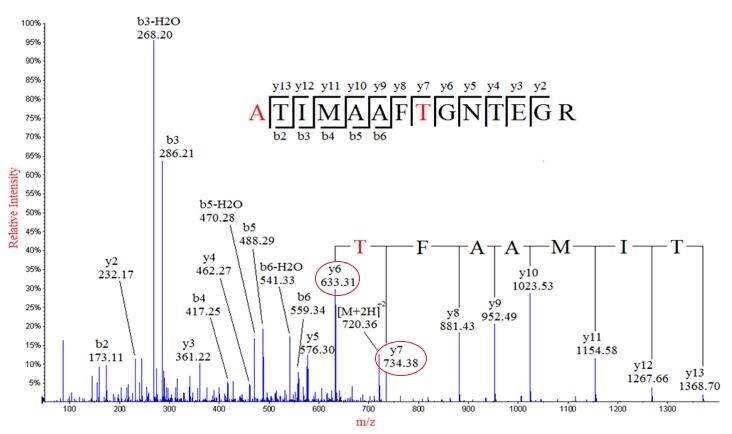
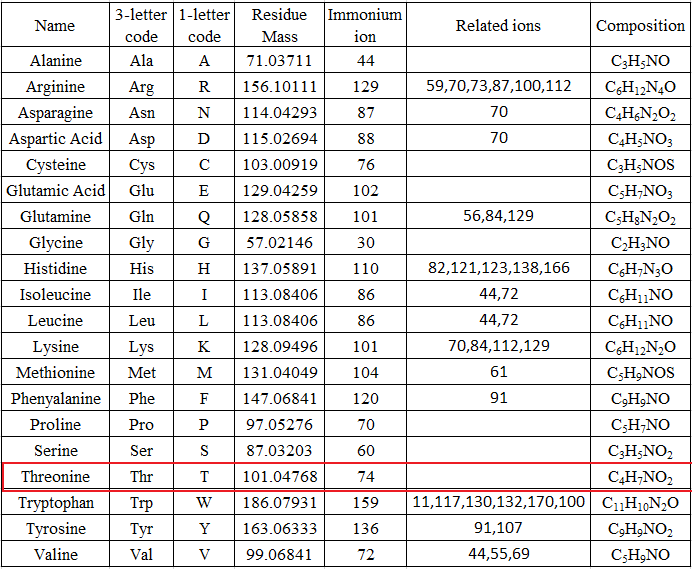
De novo sequencing can identify previous unknown peptide sequences. In addition, it can search for posttranslational modifications or for identifications of mutations by homology-based software. However, de novo sequencing will not be able to derive a complete sequence or will have uncertainty in a portion of the derived sequence. And sometimes it can be difficult to determine the directionality of a sequence. Low mass accuracy fragment ion measurements cannot distinguish between lysine and glutamine (differ by 0.036 Da) nor between phenylalanine and oxidized methionine (differ by 0.033 Da).
Equipped with state-of-the-art facilities, Creative Proteomics can provide reliable protein identification services in an affordable manner. Our team of experts with extensive experience can help you understand what you are trying to investigate and give you the most appropriate solutions. Our de novo peptide sequencing services include:
References
1. Coon J J. Collisions or electrons? Protein sequence analysis in the 21st century. 2009.
2. Ma B, Johnson R. De novo sequencing and homology searching. Molecular & cellular proteomics, 2012, 11(2): O111. 014902.