Proteomics studies play an increasing role in the field of biology. The use of mass spectrometry (MS) in combination with a range of separation methods is the main principal methodology for proteomics. The two principal approaches to identifying and characterizing proteins using MS are the "bottom-up", which analyze peptides by proteolytic digestion, and "top-down", which analyze intact proteins.
What is bottom-up proteomics?
Bottom-up proteomics involves in the proteolytic digestion of proteins before analysis by mass spectrometry. The term bottom-up implies that information about the constituent proteins is reconstructed from individually identified fragment peptides.
For the flowchart of bottom-up proteomics, by the gel approach, the protein mixture is separated by two-dimensional electrophoresis. After spot visualization, proteins are extracted from the gel, digested, and analyzed by mass spectrometry for further identification by database searching. By the gel-free approach, the protein mixture is directly digested into peptides mixture separated by multidimensional separation methods. Peptides are then analyzed by mass spectrometry. Proteins are identified from the generated mass spectra using database searching.
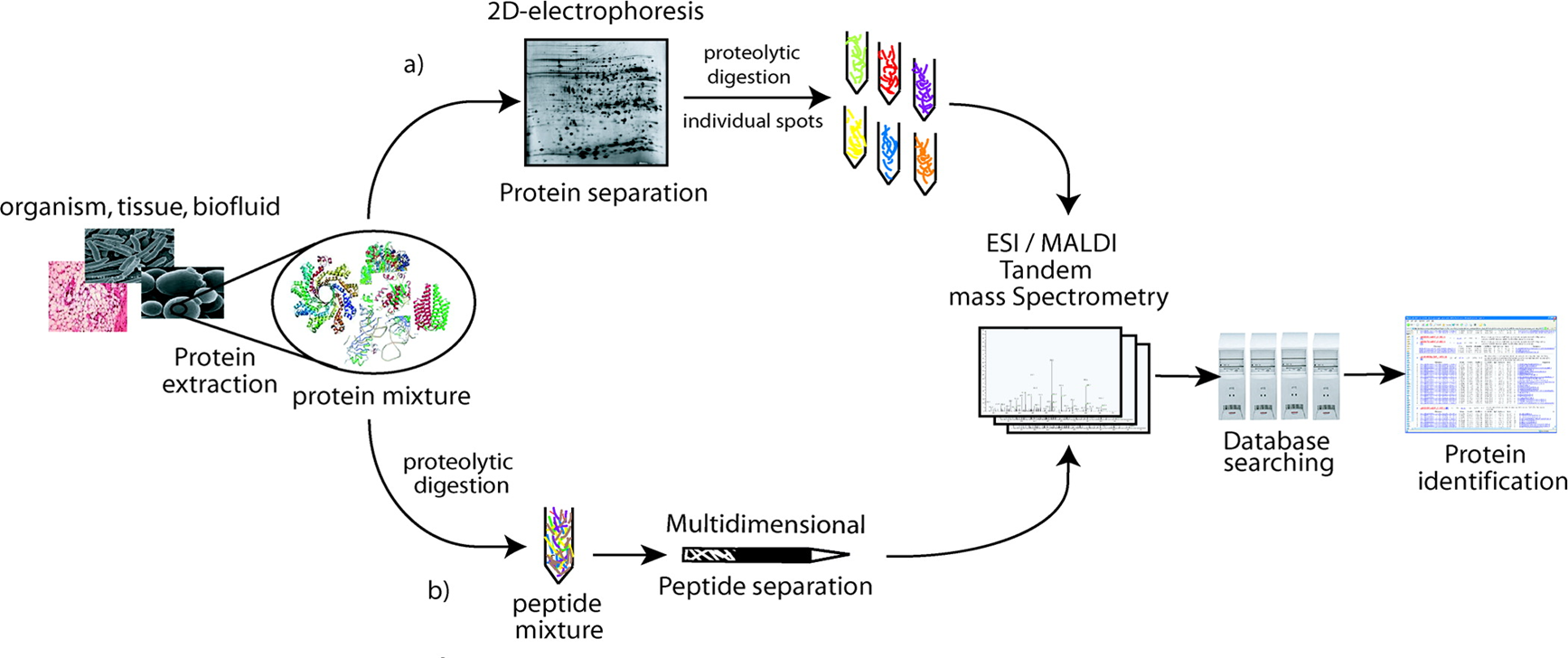
Figure 1. The flowchart of bottom-up proteomics (Fournier M. L., 2007)
Bottom-up proteomics is the most mature and most widely used approach for protein identification and characterization. It doesn’t need sophisticated instrumentation and expertise. And it has the ability to achieve high-resolution separations. There are several limitations on bottom-up proteomics. A low percentage coverage of the protein sequence is obtained because only a small and variable fraction of the total peptide population of a protein can be recovered. Genomic studies have shown that due to alternative splicing products and differences in the locations and types of post-translational modification (PTM), each open reading frame can give rise to several protein isoforms. In bottom-up proteomics, the limited sequence coverage means a significant amount of information about PTMs and alternative splice variants is lost.
What is top-down proteomics?
Top-down Proteomics can characterize intact proteins from complex biological systems. This approach routinely allows for nearly 100% sequence coverage and full characterization of proteoforms, the specific molecular form of the protein resulting from combinations of genetic variation, alternative splicing, and post-translational modifications. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation (ECD) or electron-transfer dissociation (ETD). Proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance or quadrupole ion trap mass spectrometer.
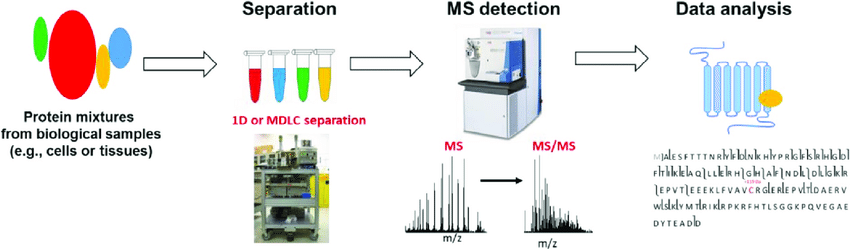
Figure 2. The flowchart of bottom-up proteomics (Lindon J. C., 2016)
It involves identifying proteins in complex mixtures without prior digestion into their corresponding peptide species. The schematic workflow for top-down proteomics includes protein separation, mass spectrometer detection, and data analysis. The main advantages of the top-down strategy are the potential access to the complete protein sequence and the ability to locate and characterize PTMs. It also has the ability for protein isoform determination. In addition, the time-consuming protein digestion required for bottom-up methods is eliminated. While there are some disadvantages.The high costs play a crucial role in the implementation of top-down proteomics in worldwide. The favored instrumentation is expensive to purchase and operate. And the favored dissociation techniques (ECT, ETD) are low-efficiency processes requiring long ion accumulation, activation, and detection times. It has not been achieved on a large scale due to a lack of intact protein fractionation methods that are integrated with tandem mass spectrometry
The comparison between bottom-up proteomics and top-down proteomics
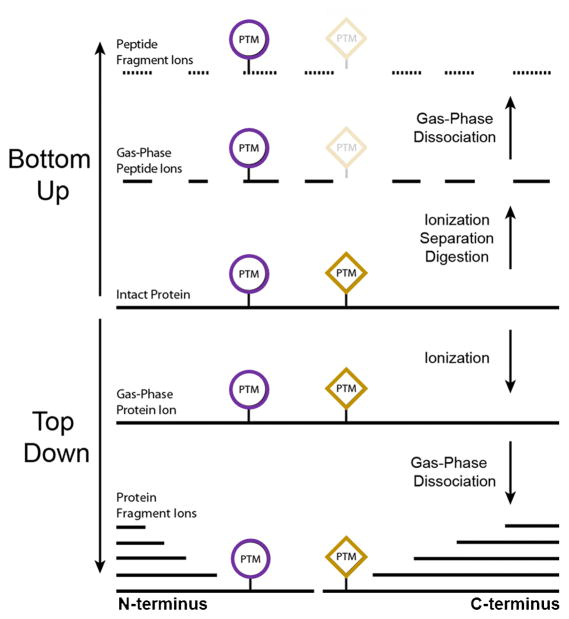
Figure 3. The comparison between bottom-up proteomics and top-down proteomics (Catherman A. D., 2014)
The bottom-up approach provides an indirect measurement of proteins through peptides derived from proteolytic digestion of intact proteins. Intact proteins are digested into peptides prior to introduction into the mass spectrometer where they are then detected and fragmented. In contrast, the top-down method measures the intact proteins. The protein are extracted from cell or tissue lysates, separated by either gel or LC, and directly analyzed by MS for a complete view of all proteoforms including those with PTMs and sequence variations. Both approaches are associated with various advantages and limitations. You should choose the appropriate approach depending on your needs
References:
- Fournier M L, Gilmore J M, Martin-Brown S A, et al. Multidimensional separations-based shotgun proteomics. Chemical reviews, 2007, 107(8): 3654-3686.
- Lindon J C, Tranter G E, Koppenaal D. Encyclopedia of spectroscopy and spectrometry. Academic Press, 2016.
- Catherman A D, Skinner O S, Kelleher N L. Top down proteomics: facts and perspectives. Biochemical and biophysical research communications, 2014, 445(4): 683-693.